Featured Article
As high-performance liquid chromatography (HPLC) celebrates its 50th year (its golden anniversary), it is appropriate to look back at its beginnings and understand the revolutionary breakthroughs that occurred as well as the ongoing evolution of this powerful separations technique. There is no doubt that HPLC has had a profound effect on analysis in virtually every field of chemistry. The column is still considered the “heart” of the system, and the role of the column throughout the history of HPLC is the focus of this article. Interestingly, the 50-year history of American Laboratory parallels the development of the HPLC technique. In fact, in my career in writing about HPLC column technology, my very first technical reviews on column developments were published in AL in the early 1970s.1,2
When did HPLC begin?
Generally given credit for the discovery of chromatography, the Russian botanist Mikhail Tswett,3 in his study of plant pigments, used liquid-adsorption column chromatography with calcium carbonate as adsorbent and petroleum ether/ethanol mixtures as eluent to separate chlorophylls and carotenoids. The technique lay dormant for years, but a number of researchers, including Martin and Synge,4 rediscovered the technique. These authors used it in their work for the separation of amino acids, inventing the technique of partition chromatography along the way. They were awarded the Nobel Prize in 1952 for their pivotal work in separations chemistry. Forward thinking, in their 1941 publication, they pointed the way for future column development with these few words: “[T]he smallest HETP (height equivalent to a theoretical plate) should be obtainable by using very small particles and a high-pressure difference across the length of the column.” It would take several decades for these predictions to be realized.
The question arises: What was the pivotal event (or perhaps series of events) that caused LC to break away from its older practice and become a technique that chemists and biologists seriously considered for their analytical results? The first is a little-known 1965 paper by Piel,5 written as a short contribution to Analytical Chemistry, where the author slurry packed finely ground silica, calcium carbonate, or alumina particles into 1- or 2-mm-i.d. glass columns. The mobile phase was driven by centrifugation or with a high liquid pressure differential using a pump capable of 3500-psi operation. The particles used varied from less than 1 μm to 0.012 μm. Centrifugal driving force was 1000–1500 times gravity and applied for five minutes while the pump’s full pressure was applied. The beds varied from 1.6 to 4.0 cm in length. Samples included various dyes as well as a spinach extract. Separations took only a few minutes for both operations. Piel claimed “excellent resolution and high capacity of microparticulate beds” and showed the results in his accompanying figures.
The second paper that was a precursor to HPLC was that of Hamilton,6 published in 1966, and applied to amino acid analysis using a pump to drive gradient solvent mixtures through the analyzer. In his work, Hamilton used 10-μm spherical ion-exchange resins. Not only did the use of the smaller, narrow particle-size distribution resin give more rapid separations compared to earlier work in this area, but Hamilton’s studies also provided insight into the effect of particle size on efficiency and phase chemistry on selectivity, things we still talk about today.
However, in my opinion, the Ph.D. research of the late Csaba Horváth, who, as a graduate student in the laboratory of István Halász at the University of Saarbrucken, Germany, developed the concept of a pellicular particle (now called a superficially porous particle, SPP) around 1968, the historical year of the foundations of HPLC.7,8 The pellicular packing, originally developed for gas chromatography, was the first LC column packing material that resulted in significant efficiency and gain in speed over the gravity feed open columns packed with large porous silica and alumina particles that were popular in the 1950s and early ’60s.
What is an SPP? Early SPPs consisted of a 45–60 μm glass bead coated with a thin (1–2 μm) porous layer of adsorbent or ion-exchange resin material. Compared to the larger porous particles with their deep porous structure, due to the more rapid mass transfer into/out of the SPP’s porous layer, these beads showed a major improvement in column efficiency while allowing users to dry-pack them into long (50–100 cm) stainless steel columns. On the downside, compared to the large-particle porous packings of yesteryear, the small surface area or resin layer provided limited sample capacity, so the columns were easily overloaded. However, separation times that used to be hours with the old columns were now tens of minutes. Higher-pressure pumps could be used to flow solvents at a faster flow rate, and a new breed of small flow cell volume ultraviolet absorption detectors allowed the improved separation power
of the SPP materials to be maintained.
However, the predictions of Martin and Synge were still to be realized and, in the early ’70s, small totally porous particles (TPPs, 5–10 μm) of silica gel became available and high-pressure slurry techniques used to pack them into narrow-bore (2–4.6 mm-i.d.) columns were published.9,10Figure 1 shows the first chromatogram obtained on a 5–10-μm TPP silica-gel column, which is quite acceptable even by today’s standards. These columns not only showed better sample capacity, but also an improved column efficiency, allowing additional separation speed to be achieved.
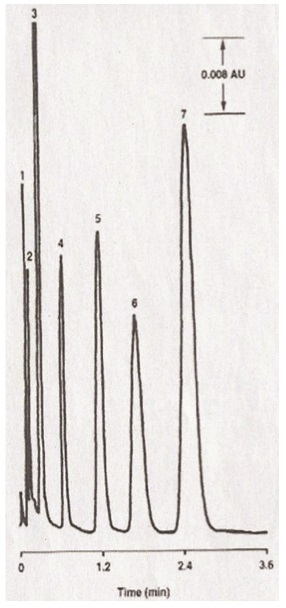 Figure 1 – Separation performed using a microparticulate silica-gel column. Column: 15 cm × 2.1 mm; mobile phase: n-hexane; flow rate: 6.6 mL/min, detector: UV absorbance; temperature: ambient; sample: 4 μL of mixture of each component. Peaks: 1) impurity; 2) phenetole; 3) nitrobenzene; 4) methylbenzoate; 5) carbazole; 6) acetophenone; 7) 2,4-dinitrobenzene. (Reprinted with permission from
Figure 1 – Separation performed using a microparticulate silica-gel column. Column: 15 cm × 2.1 mm; mobile phase: n-hexane; flow rate: 6.6 mL/min, detector: UV absorbance; temperature: ambient; sample: 4 μL of mixture of each component. Peaks: 1) impurity; 2) phenetole; 3) nitrobenzene; 4) methylbenzoate; 5) carbazole; 6) acetophenone; 7) 2,4-dinitrobenzene. (Reprinted with permission from Analytical Chemistry
, Ref. 9, 1972, American Chemical Society.)As can be seen in Figure 2, throughout the next decades, the average particle size of spherical porous packings continued to decrease, with subsequent gains in separation speed and efficiency. For many years, the most popular average particle size was 5 μm, and column dimensions of 4.6 mm i.d. × 150 and 250 mm in length were standard in most laboratories. In the 2000s, smaller TPPs became more fashionable and column efficiency showed measurable gains as did the column backpressure.
 Figure 2 – History of commercial HPLC particle development.
Figure 2 – History of commercial HPLC particle development.To keep up with the narrower peaks and the higher pressure, HPLC instruments and pumps became more sophisticated and pressures became greater. When particles became smaller than 2 μm diameter in 2003,11 backpressure greatly increased with these smaller particles since pressure increases with the inverse square of particle size (Figure 3). Thus, when the particle size is cut in half, pressure increases by a factor of 4. Pumps had to be developed with even greater output pressure. With greatly increased pressure, a new terminology arose: ultra-HPLC (or UHPLC). Finally, UHPLC pumps with pressure capability of up to 19,000 psi (1260 bar) came onto the market. In addition to higher pressure, the instruments had to be compatible with the extremely high efficiencies of these sub-2-μm columns and expanded range of flow rates, lower extra-column dead volumes, smaller injector and detector volumes, higher-speed data analysis systems, more sophisticated gradient performance became synonymous with UHPLC. Note that in the rest of this article, when the abbreviation HPLC is used, assume that UHPLC is also covered.
 Figure 3 – Comparison of different particles. Totally porous silica versus superficially porous silica.
Figure 3 – Comparison of different particles. Totally porous silica versus superficially porous silica.To illustrate the advantages of using smaller particle sizes in packed columns, Figure 4 shows a separation of steroids in three different totally porous particle size columns with decreasing column length. As noted in the figure, a decrease in particle size allows a decrease in length and still allows adequate resolution to be achieved. A decrease in length at the same flow rate results in a time savings proportional to length (assuming the column radius is the same). Higher flow rates result in even more time savings. So, in modern analytical HPLC with sub-2-μm particles, for relatively simple separations, column lengths of 30–50 mm with a 2.0–4.6-mm diameter are becoming favorites.
 Figure 4 – Reduced analysis time using 1.8-μm particles in short columns.
Figure 4 – Reduced analysis time using 1.8-μm particles in short columns.As the average particle diameters became smaller, a number of new problems arose. High backpressure placed more stress on the packed bed, and improved particle strength and better slurry packing techniques were required. More importantly, at higher flow rates, frictional heating of the mobile phase occurs as the mobile phase molecules passed through the tiny particles, causing uneven temperature profiles to occur within the column itself, especially for the wider-bore 4.6-mm-i.d. stainless steel columns. These temperature effects can cause problems with retention time reproducibility. Smaller-internal-diameter columns such as 2.1-mm-i.d. became more important since they offer better heat dissipation than larger-bore columns. In addition, mass spectrometry became a more important detection and measurement technique and the smaller i.d. columns provided less solvent to the MS source, allowing it to perform more effectively.
Reintroduction of the SPP concept
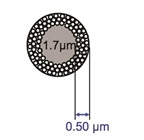
In the mid-2000s, there was a renewed interest in the SPP concept, mainly based on the work of pioneer J.J. Kirkland.12 Rather than the large glass bead of yesteryear, this new breed of packing used a much smaller (1.7-μm-diameter) solid-core silica bead as the base material (Figure 3). A thicker layer of silica (0.5 μm) was coated onto the core material, making the entire particle 2.7 μm in diameter. Due to the smaller particle, limited diffusion path, and denser packing, the chromatographic performance improved enough to provide efficiency as good as the sub-2-μm porous particles that had spurred the UHPLC interest. Figure 5 depicts the same sample on a TPP and an SPP column with similar stationary phase chemistry run under the same chromatographic conditions. Note that the separation is nearly the same, but the pressure is considerably lower for the SPP column. Since the particle diameter was larger, the pressure drop decreased relative to the sub-2-μm TPP particle, which would allow longer columns and faster flow rates to be used. Since their introduction, both larger (up to 5 μm) and smaller (down to 1.3 μm) SPP columns with varying shell thicknesses have been commercialized. Wide-pore silica (pore diameter 300–400 Angstroms) have also become available for the rapid separation of larger molecules such as proteins.
 Figure 5 – Comparative separations on a totally porous 1.8-μm particle (TPP) and 2.7-μm superficially porous particle (SPP).
Figure 5 – Comparative separations on a totally porous 1.8-μm particle (TPP) and 2.7-μm superficially porous particle (SPP).Some other side benefits of using SPP instead of TPP have arisen. For example, heat dissipation appears to be better on these solid-core materials and thus frictional heating seems to be reduced.13 The denser particle packs better and thus there is a reduction in the “A” term of the van Deemter equation, which provides a large contribution to the efficiency gains of modern SPPs. A thicker shell, relative to the early SPPs, provides sufficient sample capacity for preparative separations. Simple calculation of the available porous phase for the popular 2.7-μm SPP shows that there is only a 25% reduction in total surface area, relative to a totally porous particle of the same size, due to the thicker shell. The reduction of pressure requirements for SPP relative to TPP has limited the pressure race in modern HPLC. Pressures in the thousand-bar range are no longer required for the bulk of separations. So, it is no wonder that there is a great deal of interest in modern SPP columns. Many laboratories are developing their new methods on this type of column, and the future of SPP looks very bright.
Other areas of column development
Monoliths
Monolithic columns do not consist of individual particles but of a continual network of silica or polymer phase that contain two types of pores: flow-through pores with macroporosity (1–2 μm in width) and diffusive pores called mesopores. The mesopores are those that contain bonded-phase moieties that control the separation mode. Because of the flow-through pores, the monoliths show very low pressure drops, less than half that of a TPP column with the same efficiency and lower than SPP columns of the same dimensions.
It turns out that monoliths have been around for some time, introduced several decades ago by Bio-Rad Laboratories as UNO columns. Never very well-known, these polymeric monoliths are only available with ion-exchange functionality and are recommended for bioseparations. They were never adopted by HPLC advocates. Instead, in the late ’90s, BIA Separations of Ljubljana, Slovenia productized macroscopic monolithic columns in a disk format and applied them for the separation of biomolecules, namely proteins. With functionalities such as DEAE ion exchanger and Protein A affinity and other popular phases, BIA focused its efforts mainly on the preparative and process industry and less so on the analytical size market. Isco, now part of Teledyne, licensed the patents of Cornell University based on the technology developed by Svec and Fréchet14 based on rigid macroporous polymer monoliths and began to produce Swift monolithic columns consisting of polystyrene-divinylbenzene functionality. Eventually, Teledyne spun off the product line and sold it to Dionex/Thermo Fisher, which now markets these columns.
Polymeric monoliths have been studied extensively for the last 20 years or so, and each HPLC series meeting leads the pack in column technology presentations with researchers developing new approaches to increase their application potential. Originally, polymeric monoliths were thought to be applicable only to large molecules, but recent work on polymerization techniques has found application to small molecules as well. Unfortunately, very few of these research efforts have led to commercial products that chromatographers can use to solve their separation problems. These polymeric monoliths have the advantage of a wide range of pH compatibility, high loading capacity, and use at high flow rates at low pressures. Yet, only a few products are on the market and few new ones are introduced each year.
The first-generation silica monoliths were developed by the laboratory of Nakanishi and Tanaka at the Kyoto Institute of Technology in Japan.15 Commercially, they were first introduced by Karin Cabrera of EMD Millipore at HPLC ’98 and at Pittcon as SilicaRod columns in 2000.16 Later, when new dimensions were developed, the name was changed to Chromolith and a second-generation silica monolith followed later. Unlike polymeric monoliths that can be produced in situ inside the column, silica monoliths are produced in rod-like structures that shrink as the synthesis progresses. Thus, the silica rods have to be cladded into a polymeric sleeve before they can be connected to a pump and detector. In 2001, Merck scientists perfected the technique to tightly clad the silica rod with PEEK polymer, thereby eliminating well effects and allowing smooth flow down the column matrix. Unfortunately, there were two problems with the technique: 1) rod lengths were limited to 150 mm and 2) the PEEK enclosure limited the column to a maximum pressure of 125 bar. Thus, to get high efficiency, columns had to be coupled and, since a single monolithic column was already expensive, multiple columns were even worse, compared to conventional packed analytical columns.
A unique feature of silica-based monolithic HPLC columns over packed particle columns is the ability to independently control the macro- and mesopore sizes as well as the silica skeleton size. This control is achieved through the sol-gel preparation process; as a result, this process permits the design of HPLC columns that show simultaneous high separation efficiency and high permeability, not entirely possible with most packed particle columns. In the latter case, the particle size determines the separation efficiency and permeability but in a reciprocal manner, i.e., big particles lead to high permeability and low efficiencies, whereas small particles lead to the opposite. The SPP columns do provide some of the advantages of high efficiency but with lower permeability than the silica monoliths.
By a careful balance of the macro- and mesopore sizes of the silica monolith, one can balance efficiency and permeability. The macropores allow flow to pass more easily through the column and control the permeability, while mesopores provide the surface area and control loading capacity and efficiency. As far as chromatographic performance, the first-generation silica monolith had about the same efficiency as a 3.5–5-μm totally porous particle.16 The big difference between the first-generation (Chromolith) and second-generation (Chromolith HR) columns is the macropore size, which is around 2 μm to 1.1 μm, respectively. Thus, Chromolith HR columns are more efficient (140,000 plates/m vs 80,000 plates/m) but have higher pressure drops (65 bar for a 100-mm × 4.6-mm column with a 60:40 [v/v] acetonitrile:water mobile phase flowing at 2 mL/min vs 25 bar for a Chromolith first-generation) due to the change in the macropore/mesopore domain ratios.16 The newest silica monoliths still permit the user to employ conventional 400-bar HPLC instrumentation. A comparison of the latest silica monolith and a typical SPP is depicted in Figure 6. Note the lower pressure drop on the monolith column.
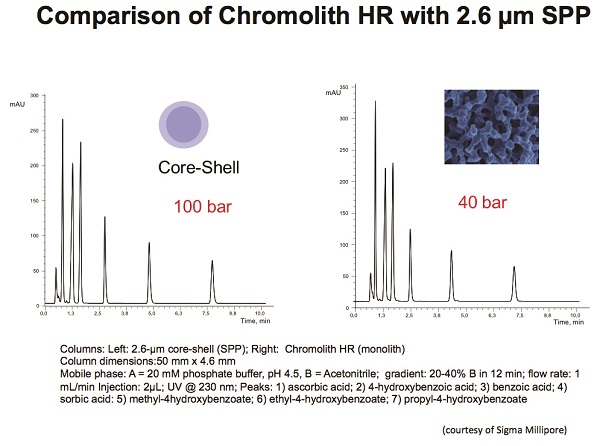 Figure 6 – Comparison of Chromolith HR with 2.6-μm SPP. (Image xourtesy of MilliporeSigma, Billerica, MA.)
Figure 6 – Comparison of Chromolith HR with 2.6-μm SPP. (Image xourtesy of MilliporeSigma, Billerica, MA.)
The downside is that this patented technology has not been made widely available, since most of the production of silica monoliths is controlled by a single supplier. This has limited the acceptance of silica-based monoliths, especially in the pharmaceutical industry, where a second source is often required. In addition, lack of competition and further research and development by other commercial companies has slowed down the potential growth of the silica monoliths. Nevertheless, the future still looks bright for monolithic columns.
For those readers who are interested in more details of polymeric and silica-based monolithic columns, Frank Svec, a leading scientist in the monolithic field, has published a series of general reviews of these technologies17-20 in addition to his book on the same subject.21
Stationary phase development
The study of particle morphology has only been part of the story. Remember, our ultimate goal in HPLC is to obtain the best possible resolution for the target compounds in the shortest possible time. The decrease in particle size and particle morphology has greatly contributed to efficiency gains in HPLC. However, it has long been known that N (efficiency) contributes only nominally to resolution. Figure 7 shows the relative impact of the three factors (k’ retention, N efficiency, and α selectivity) in the resolution equation, also depicted in the figure. Gains in efficiency, although helpful, are somewhat limited by the fact that N1/2 only produces a 40% increase in resolution for a doubling of efficiency. The impact of k’ is even smaller but, of course, retention is needed to achieve a separation. Clearly, if one could influence α, it would have the most dramatic impact on resolution.
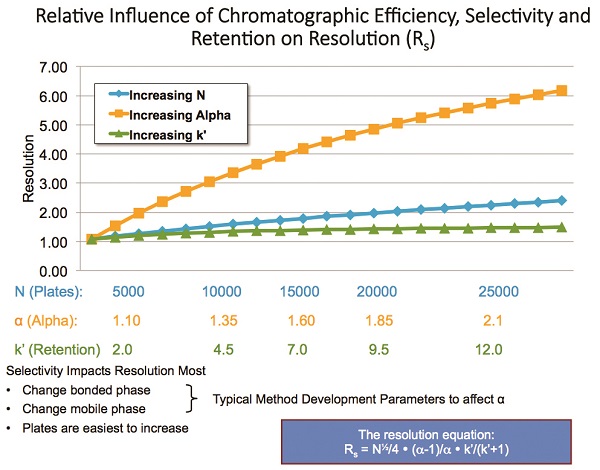 Figure 7 – Relative influence of chromatographic efficiency, selectivity, and retention on resolution (Rs).
Figure 7 – Relative influence of chromatographic efficiency, selectivity, and retention on resolution (Rs).The earliest stationary phases used in HPLC with the large particle SPP (then called pellicular packings) were coated phases. The packing would be either coated externally and the particles dry-packed or the coatings were applied in situ. The dominant form of liquid chromatography was liquid–liquid chromatography, where analytes would partition into the coated phase. Unfortunately, if extreme care was not exercised, the coatings could easily be stripped from the packed bed and retention times drifted. The mobile phase had to be saturated with stationary phase, gradient elution wasn’t feasible, and the column temperature had to be strictly controlled to prevent retention changes. It wasn’t long before researchers developed chemically bonded phases that would negate some of the disadvantages of the coated phases. Early chemical bonded phases of the brush type with Si-O-C bonds were easily hydrolyzed and thus could not tolerate any water or other proton-donating liquids. Siloxane (Si-O-Si-C) bonded phases proved to be more stable and could be used with a wide variety of solvents and pH values. Even today, siloxane phases are the most popular and are available with a wide variety of chemistries.
The availability of bonded siloxane phases had a particularly favorable fallout—the development of reversed-phase liquid chromatography (RPLC). The earliest stationary phases in HPLC were developed for adsorption and normal-phase chromatography. These stationary phases were polar (e.g., silica gel, β,β’ oxydipropionitrile) and the mobile phase was nonpolar (e.g., isooctane, hexane). The elution order was nonpolar compounds first, followed by compounds of increasing polarity. This was the “normal” practice of HPLC initially. In the new mode of RPLC, never very popular in liquid–liquid chromatography, the opposite combination of phases from the normal operation was used. In RPLC, the stationary phase was nonpolar (e.g., octadecylsilane, octylsilane) and the mobile phase was polar (e.g., water and water-miscible organic solvents like methanol and acetonitrile). Hence, the name (coined by Csaba Horváth) was reversed-phase, a name still used today and, by far, the most popular mode of HPLC/UHPLC.
Almost from the beginning of bonded-phase packings, RPLC exploded, especially during the ’70s, and has been widely used ever since. RPLC not only can separate nonpolar from polar compounds but can also separate ionic and ionizable compounds by adjustment of the pH via buffers. Many compounds that show low solubility in water and higher solubility in water-miscible organic solvents are amenable to RPLC; over the years it has even increased in popularity. Not only are popular alkylsilane C8 and C18 phases available, but a wide variety of other phases based on cyclic alkyl, aryl, mixed aryl-alkyl, fluorinated, and polar-embedded phases as well as aryl or alkyl ion-exchange mixed-mode phases have found some niche applications. In a 2010 publication,22 I accounted for over 1500 reversed-phase columns in more than 170 different chemistries that have been introduced during the 1970–2010 timeframe. Over 92% of all liquid chromatographers use RPLC at some time in their laboratory.22
Since RPLC mainly relies on hydrophobic interactions, selectivity changes are moderate going from one of these phases to another. In order to achieve larger α values, one has to almost certainly change the LC mode. Besides RPLC, the popular modes in HPLC are: 1) normal-phase, where the stationary phase is polar (e.g., silica, diol) and the mobile phase is nonpolar (e.g., hexane, dichloromethane); 2) ion-exchange, where the stationary phase shows cationic or anionic functionality and large selectivity differences are noted for weak and strong ionogenic functional groups; 3) size exclusion chromatography (SEC), where ideally there is no interaction between the analyte and the packing material (pure diffusion into controlled-size pores is the governing mechanism); 4) chiral chromatography, where chiral compounds interact with chiral centers on the packing material, thereby separating R- and S-enantiomers; and, last but not least, 5) hydrophilic interaction liquid chromatography (HILIC), which resembles RPLC utilizing the same mobile phases but with high concentrations of organic solvent and relies upon analytes, mostly polar, to partition into a thin film of water on the polar stationary phase.
Unfortunately, selectivity is very difficult to predict and thus it is difficult to generate a specific phase based on scientific principles and know the exact separation behavior of that phase. So, until more study is conducted, we often have to rely on “trial-and-error” experiments to determine if additional or different selectivity is achieved.
Other chromatographic media and stationary phase chemistry
Although silica gel is, by far, the most commonly used LC medium, in the last 25 years, a number of other sorbents has been investigated for use in LC columns. Polymers have long been employed as chromatographic packings either as pure polymer (e.g., polystyrene crossed linked with divinylbenzene, PS-DVB) or as functionalized PS-DVB (e.g., sulfonated for ion exchange or derivatized with alkyl chains to resemble reversed-phase media). Other polymers such as polymethacrylate, polyacrylamide, and divinylbenzene also find use as HPLC base materials. Polymers, for the most part, display a much wider pH range than silica-gel materials, but suffer from a lack of column efficiency and, depending on their degree of crosslinking, may swell or shrink with changes in the mobile phase. Due to their ruggedness, over the years, silica-gel-based ion exchangers have given way to polymers where strong buffers, high pH, and higher temperatures are often encountered. Ion chromatography strictly uses polymer-based media. Polymer-based ion-exchange phases with strong- and weak-ionic and ionizable functionalities are widely used in the proteomics and genomics separation area as well as for the separation of other ionic and ionizable organic and inorganic compounds, particularly in the area of ion chromatography. Separations of carbohydrates at high pH with pulsed amperometric detection make use of polymeric ion chromatography packed columns.
Nonaqueous size-exclusion chromatography (SEC) is also dominated by polymeric media, where pore sizes can be easily varied. In recent years, new synthesis techniques have allowed nonaqueous SEC columns to undergo solvent changes without undue swelling and shrinkage. For aqueous SEC, silica-gel-based columns do find use. Smaller-particle SEC columns have recently become available, allowing large-molecule characterizations that used to take hours be performed in several minutes.
Other inorganic media introduced over the last 25 years include zirconia, titania, and graphitic carbon, mostly as niche products, but these have never replaced silica as the mainstay in HPLC column packing.
Over the years, improvements have been made in the base silicas used for bonded phases, including the development of Type B low trace metal, nonacidic packings, providing better separations and improved peak shapes. Just about every new column introduction made is based on a Type B silica; Type A silicas are now part of history. Exhaustive endcapping was another procedure to remove silanols, which cause tailing under certain mobile phase conditions. Silica-organic hybrids such as Xterra and bridged-ethyl hybrid (BEH) from Waters23 offer wider pH capability and can also withstand the high pressures of UHPLC.
Two of the most exciting areas of stationary phase development over the past 25 years were the advent of chiral phases and phases for HILIC. The former phases were quickly accepted in the pharmaceutical industry, since many of the drugs under development have one or more chiral centers and regulatory bodies require the analysis of both enantiomers. In some cases, one of the enantiomers has detrimental side effects for patients, while the other enantiomer provides the drug function. One of the highlights in chiral column development was the 2005 introduction of immobilized phases, allowing for more rugged columns.24 The introduction of chiral phases for supercritical fluid chromatography (SFC) gave a rebirth to this technique, initially for preparative and process applications, but now for more general-purpose chromatography. Introduction of a new generation of SFC instrumentation has spurred the resurgence. For details on developments in chiral phases in recent years, see the reviews of Beesley25-29 and Ward and Ward.30,31
Developed many years ago by Andy Alpert,32 HILIC laid dormant for a number of years. It is a separation technique for highly polar analytes that gets around some of the problems associated with reversed-phase chromatography such as low (or no) retention of polar analytes or phase collapse (phase dewetting). HILIC columns often have the opposite effect from RPLC in that polar analytes generally elute later than nonpolars. Many pharmaceutical compounds contain polar functional groups such as amine and carboxylic acid, so HILIC has found widespread use in this marketplace. It uses a polar stationary phase such as bare silica gel, polar bonded phase (e.g., diol), and certain mixed-mode or zwitterionic phases. Operating conditions usually require a high percentage of a nonpolar mobile phase, similar to the requirements for normal-phase chromatography. However, unlike normal-phase, which uses nonpolar solvents like hexane and methylene chloride and tries to exclude water from the mobile phase, HILIC requires some water (~2%) in the mobile phase to maintain a stagnant enriched water layer on the packing surface into which analytes may selectively partition. In addition, water-miscible organic solvents are used.
Under HILIC, polar analytes are well retained and elute in order of increasing hydrophilicity. HILIC is especially favored by mass spectroscopists since ionization efficiency is often enhanced in organic solvents and the presence of lower concentrations of volatile buffer salt compared to RPLC. Figure 8 cites the work of Guo and Gaiki and shows considerable selectivity differences between four silica-based HILIC phases—aminopropyl, amide, zwitterionic, and bare silica—for the separation of some acidic solutes. They attributed increased retention and different selectivity on the aminopropyl phase to ionic interactions. The improved resolution of aspirin and 4-aminosalicylic acid, compared with that on a bare-silica phase, was suggested to be due to electrostatic interaction between the acids and the negatively charged sulfonate groups on the sulfoalkylbetaine phase.
 Figure 8 – Separation of acidic compounds on four different HILIC columns. (Image courtesy of Y. Guo and S. Gaiki, Johnson and Johnson, Raritan, NJ.)
Figure 8 – Separation of acidic compounds on four different HILIC columns. (Image courtesy of Y. Guo and S. Gaiki, Johnson and Johnson, Raritan, NJ.)Conclusion
To close, HPLC/UHPLC columns have been brought to the forefront of separations technology. Column reproducibility has improved, column stability/lifetime has been increased, convenient connectivity has been mastered, efficiency has increased by orders of magnitude, a wide variety of regular and specialty stationary phases have brought difficult separations into the routine lab, and cost/injection has decreased
References
- Majors, R.E. Am. Lab. 1972, 4(5), 27–38.
- Majors, R.E. Am. Lab. 1975, 7(10), 13–36.
- Tswett, M.S. Proceedings of the Warsaw Society of Naturalists, Biology Section, 1905,14(6), 20–39.
- Martin, A.J.P. and Synge R.L.M. Biochem. J. 1941, 35, 1358–68.
- Piel, E. Anal. Chem. 1965, 38(4), 670–2.
- Hamilton, P.B. In Advances in Chromatography, Giddings, J.C. and Keller, B., Eds. Marcel Dekker: New York, NY, 1966), Vol. 2, p. 3.
- Halász. I. and Horváth, C. Anal. Chem. 1966, 36, 1178.
- Horváth, C.; Preiss, B.A. et al. Anal. Chem. 1967, 39, 1422.
- Majors, R.E. Anal. Chem. 1972, 44(11), 1722–6.
- Kirkland, J.J. J. Chromatogr. Sci. 1972, 10, 593–9.
- Majors, R.E. LC·GC N. Amer. 2003, 21(3), 240–59.
- Kirkland, J.J. Anal. Chem. 1992, 64, 1239–45.
- de Villiers, A.; Lauer, H. et al. J. Chromatogr. A 2006, 1113(1–2), 84–91.
- Svec, F. and Fréchet, J.M.J. Anal. Chem. 1992, 54, 820.
- Tanaka, N.; Ishizuka, N. et al. Kuromatogurafi 1993, 14, 50.
- Cabrera, K. LC·GC N. Amer. 2013, 30(S4), 30–5.
- Svec, F., LC·GC N. Amer. Recent Developments in LC Column Technology, June 2004, 18–21.
- Svec, F. and Geiser, L. LC·GC N. Amer. 2006, 24(S4), 22–7.
- Svec, F. and Krenkova, J. LC·GC N. Amer. 2006, 26(S4), 24–30.
- Svec, F. LC·GC N. Amer. Recent Developments in LC Column Technology, Apr 2010, 18–23.
- Svec. F.; Tennikova, T.B. et al., Eds. Monolithic Materials: Preparation, Properties, and Applications. Elsevier: Amsterdam, 2005.
- Majors, R.E LC·GC N. Amer. Recent Developments in LC Column Technology (special supplement), Apr 2010, 8–17.
- Wyndham, K.; Walter, T. et al. LC·GC N. Amer. 2012, 30(S4), 20–9.
- Beesley, T.E. LC·GC N. Amer. 2006, 24(S4), 28–31.
- Beesley. T.E. and Lee, J.T. LC·GC Eur. 2003, 16(6a), 33–6.
- Beesley, T.E. and Lee, J.T. LC·GC N. Amer. Recent Developments in LC Column Technology, June 2004, 30–3.
- Beesley, T.E. LC·GC N. Amer. 2006, 24(S4), 28–31.
- Beesley, T.E., LC·GC N. Amer. 2008, 26(S4), 43–6.
- Ward, T.J. and Ward, K.D. LC·GC N. Amer. 2012, 30(S4), 43–5.
- Ward, T.J. and Ward, K.D. LC·GC N. Amer. 2014, 32(S4), 20–3.
- Alpert, A.J. J. Chromatogr. 1990, 499, 177–96.
- Alpert, A.J. J. Chromatogr. 1990, 499, 177–96.
Ronald E. Majors is a chromatography consultant whose company is ChromPrep; e-mail: [email protected]