Featured Article

Far from being a weed with “roots in hell,” as portrayed during 1930s propaganda campaigns, cannabis could well be a wonder drug, and the next target of precision medicine, with strains selected for the specific needs of individual patients.
Medicinal cannabis is highly valued for its pharmacologically active cannabinoids, primarily found in the resin produced in the flowers of female plants. It also contains a variety of monoterpenes and sesquiterpenes, and differences between the pharmaceutical properties of different cannabis strains have been attributed to interactions between these compounds.1 In fact, there are more than 483 different identifiable chemical constituents known to exist in Cannabis sativa, of which over 80 are unique to the cannabis plant. These include nitrogenous compounds, amino acids, proteins, glycoproteins, enzymes, sugars and related compounds, hydrocarbons, alcohols, acids, esters, aldehydes, ketones, fatty acids, lactones, steroids, terpenes, noncannabinoid phenols, flavonoids, vitamins, pigments, and other elements.
The toxicology of cannabinoids is remarkable—cannabinoids are extremely safe and well-tolerated. Heat converts cannabinoid acids to neutral molecules that bind to endocannabinoid receptors found in the vertebrate nervous system. This pharmacological activity leads to analgesic, antiemetic, and appetite-stimulating effects, and may alleviate symptoms of neurological disorders. Terpenes are believed to contribute anxiolytic, antibacterial, anti-inflammatory, and sedative effects.
Among the two most abundant natural derivatives of cannabis are tetrahydrocannabinol (THC) and cannabidiol (CBD). The intoxication or euphoria (the “high”) that comes from ingesting marijuana is derived from THC, whereas CBD does not have psychoactive effects, and is already being incorporated into accepted therapies, including the FDA-approved drug for the treatment of a rare form of epilepsy (Epidiolex, Greenwich Biosciences, Carlsbad, CA).
Breeding based on quantities of these compounds is vital to the valuable industry that is growing in light of increased state legalization of cannabis for both medicinal and recreational use. Yet it is extremely difficult to study the genetics of this plant due to limited funding and drug prohibition, which has restricted access to cannabis by plant breeders and researchers. Until recently, the scientific and medical communities have been completely in the dark about how cannabis works.
Medicinal Genomics (Beverly, MA) first attempted to sequence the cannabis genome in 2011. At that time, the sequencing technology was not able to handle all the repeat content and the polymorphism of the genome. The diploid cannabis genome is 10 times more varied than the human genome. It is highly repetitive, comprised of 66% A and T nucleotides, with 99.5% identical cannabinoid genes arrayed across a landscape of viral long terminal repeats (LTRs) 30,000–120,000 bases in size. The draft assembly included hundreds of thousands of pieces (contigs) and was hardly a useful resource for research.
Over the next seven years, many researchers tried to improve it, but they were only achieving average contig N50 lengths of 159 kb. Recently, the Medicinal Genomics team made another attempt to sequence the cannabis genome, this time with a technology promising to provide longer sequences.
DNA isolation
The first challenge was extracting enough high-molecular-weight DNA from this hardy plant. A novel DNA isolation process developed by Medicinal Genomics—SenSATIVAx for MIP/Extract—was applied (see Figure 1). It uses magnetic particles to isolate and purify both plant and microbial DNA from a marijuana-infused product or a flower extract.
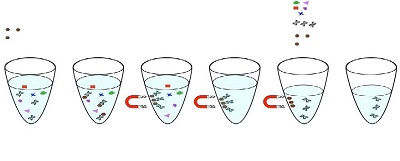 Figure 1 – Protocol for SenSATIVAx for MIP/Extract.
Figure 1 – Protocol for SenSATIVAx for MIP/Extract.Designed for ease of use and minimal requirement of laboratory equipment, the approach replaces large centrifuges with lightweight mini-fuges, magnetic particles, and magnets. The use of magnetic particles affords 8- or 96-tip automation, enabling minimal entry costs and high-throughput applications. DNA can be isolated in under 1 hour with a hands-on time of less than 45 minutes.
Several modifications to this technique were required to capture high-molecular-weight DNA ideally suited for long-read assembly. Sufficient DNA was extracted from isopropanol-treated stalk and leaf of Jamaican Lion, a Type II strain of cannabis that makes THCA and CBDA. This DNA was then sequenced using the latest version of SMRT Sequencing hardware, software, and chemistry from Pacific Biosciences (PacBio, Menlo Park, CA).
The PacBio Sequel System allows users to essentially watch a movie of a polymerase replicating DNA at a single molecule level. It delivered 32,000 base pair reads—nearly 1,000 times longer than the reads used in the 2011 draft genome.
For the Jamaican Lion assembly,2 more than 180 billion bases were sequenced on the Sequel System, and the longest reads were selected as the foundation for the DNA assembly process. These reads are so long and accurate that every base is covered over 15 times with 60,000 base pair reads. This is an order of magnitude more contiguous than anything produced to date, and of better quality than the Human Genome Project effort that was celebrated at the White House in 2001.
DNA assembly
Sequencing libraries were constructed with the help of scientists at PacBio, and 91 Gb of raw sequence data with an N50 read length of 32 kb was produced. These were assembled into contigs with an N50 length of 665 kb with the assembly tools FALCON and FALCON-Unzip.
Three PacBio assemblies were made and evaluated. In all cases, the genome size was 2–5 times larger than previous genome estimates, representing the assembly of the two distinct haplotypes in the cannabis genome.
A BLAST analysis was performed to examine the synthesis of some key compounds (THCA, CBCA, and CBDA). Prior to this assembly, the genes were always on different small contigs or not in the contigs at all. The PacBio assembly not only assembled these genes into tandem repeats, but also highlighted many other copies of mutated synthase genes clustered around each synthase gene (see Figure 2). This copy number expansion had been explored before, but never resolved into contiguous tandem repeats.
 Figure 2 – Hi-C scaffolds of cannabis synthesis gene clusters.
Figure 2 – Hi-C scaffolds of cannabis synthesis gene clusters.Previous studies have suggested that the THCAS and CBDAS genes are linked. The new, contiguous genome assembly showed these two genes to be on separate contigs, but with signs of tandem repeat structures. The researchers initially found 3–4 tandem copies (all directionally orientated) of CBCA synthase and multiple partial copies of THCA synthase, spaced 30+ kb apart and interspersed with repeat elements. Thus, longer and more accurate reads were collected in an attempt to further resolve the loci.
An additional 34 Gb of 44 kb-long reads generated on the Sequel 3.0 chemistry were collected and reassembled, resulting in a 3.8-Mb contig N50 with a 97% BUSCO completeness score. The assembly resulted in a partially phased genome of 1.07 Gb in 558 contigs, with the largest contig being 32 Mb, making it five times more contiguous than anything published to date.
Augmenting the assembly from 91 Gb of data from Sequel 2.1 chemistry, which was 32 kb, with just 34 Gb more 3.0 chemistry, had a substantial increase in the contig N50 length of the assembly (665 kb to 3.8 Mb). Due to the extra length of these raw reads, many more of the CBCA synthase genes were bridged. The accuracy to separate genes that are over 99.9% identical but separated by long terminal repeats was also critical.
Just the beginning
The above genome assembly is better than most agricultural crops, which is fitting for a plant that has the potential to be more valuable than wheat and corn combined, with far more varied and diverse uses—from CO2 sequestration to pain relief and medical therapies to complete vegetable proteins, clothing, and industrial materials.
A comprehensive cannabis reference genome will be a tremendous help in understanding the genetics of the plant and how to breed for more CBD or different esoteric cannabinoids. It opens the door to a host of industry innovations, including enabling cannabis research in states and countries where it is still illegal, marker-assisted selection for genetically based strain identification, accelerated breeding to improve production yields, reliable seed-to-sale tracking systems, and pathogen identification to ensure cannabis purity and safety. Just a few months after this public release, cannabis proteomics preprint papers3 are beginning to leverage the data.
Early analysis based on the genome has already led to insights into CBD and THC synthesis in Type II plants. An assortment of diploid alleles suggests there can be 1:2 and 2:1 functional gene dosages for CBDAS:THCAS in the diploid plant, for instance. The genome has also facilitated the development of quantitative PCR assays that can accurately quantify fungal DNA present in cannabis samples. This is important because the risk of illness due to pathogenic mold and bacteria on agricultural products extends to medical cannabis, yet current culture-based foodborne illness testing kits may not be sufficient to detect fungal threats like the carcinogenic aflatoxin produced by the endophytic Aspergillus species.
The clearance of aflatoxin requires the human liver enzyme CYP3A4, and this enzyme is potently inhibited by cannabinoids. Likewise, the spores of these fungi have been associated with fatal Aspergillosis in cannabis smokers.4 Since endophytes are difficult to extract from the plant without lysing cells, qPCR must be selective enough to amplify fungal DNA in a complex and overwhelming cannabis background, which until recently was poorly documented. The genome sequence has vastly improved primer design and assay conversion rates.
This genomic resource can also improve plant pathogen testing. Crop loss in cannabis cultivation is a big concern, as it is often grown indoors, with monoclonal plants that are highly susceptible to diseases like powdery mildew. Cannabis growers face additional challenges because cannabis is an all-cash industry, with no access to banking and, often, lack of crop insurance, due to its federal designation as a prohibited drug.
Longer and more accurate reads are making previously illegible genomes legible, without the need for utilizing traditional linkage maps across a population.
In order to fully understand the complex cannabinoid and terpene pathways, many more cultivars require sequencing. Ideally, researchers will produce a pangenome, with references for Type I, Type II, and Type III cannabis, and hemp lines, all sequenced with the same technology.
These refined cannabis genome assemblies would aid in better marker-assisted selection, refined synthetic biology programs, and meaningful QTL maps. They would also form the basis for a blockchain-secured, seed-to-sale tracking system that can ensure quality control for patients and consumers.
References
- Russo, E.B. Taming THC: potential cannabis synergy and phytocannabinoid‐terpenoid entourage effects. Brit. J. Pharmacol. 2011, 163, 1344–64.
- McKernan, K.; Helbert, Y. et al. Cryptocurrencies and zero mode wave guides: an unclouded path to a more contiguous Cannabis sativa L. genome assembly. OSF Preprints 2018; https://osf.io/gf2rv/
- Jenkins, C. and Orsburn, B. Constructing a draft map of the cannabis proteome; bioRxiv 2019; doi: https://doi.org/10.1101/577635.
- Remington, T.L.; Fuller, J. et al. Chronic necrotizing pulmonary aspergillosis in a patient with diabetes and marijuana use. CMAJ 2015, 187(17), 1305–8.
Kevin McKernan is the CSO and founder of Medicinal Genomics, Beverly, MA, U.S.A. Sarah Kingan is a staff scientist in the Bioinformatics Application Team at Pacific Biosciences (PacBio), 1305 O’Brien Dr., Menlo Park, CA 94025, U.S.A.; tel.: 650-521-8000; e-mail: [email protected]; www.pacb.com.