Mass spectrometry (MS)-based proteomics typically employs multiple sample processing steps, representing a crucial part of routine MS analyses. Complex workflows, extensive sample fractionation, and proteolytic digestion are highly time-consuming and restrict the overall technical reproducibility. The accuracy and robustness of the MS platform is also strongly affected by sample quality reasoning for high-quality proteomic samples. In 2016, PreOmics introduced the in-StageTip (iST) technology dedicated to streamlined sample processing for MS-based protein analysis and proteomics.

Figure 1: Picture of the iST kit for the 8 reactions, including cartridges, plastic ware, chemicals and enzymes.
Based on the original publication in 20141, PreOmics (Martinsried, Germany) has further optimized the iST technology to streamline protein sample processing. The iST kit is an all-in-one solution providing all chemicals, proteolytic enzymes, and polymer-free plastic ware to perform a successful sample preparation for MS-based protein analysis (Figure 1). High reproducibility (R2 >0.9) has been demonstrated over a working range of 1-100 µg protein input. While the whole workflow is performed in a single enclosed volume called cartridges (Figure 2), sample contamination and loss are minimized. Moreover, it provides a significant time advantage (>40 hours saved) compared to many common protocols.

Figure 2: Graphical abstract of the iST workflow depicting processing steps on the cartridge.
The iST technology employs a 3-step procedure (Figure 3). The first step combines cell lysis, protein denaturation, protein reduction and alkylation within 10 minutes at 95°C. The second step utilizes a mix of proteolytic enzymes to efficiently digest proteins to peptides within 60 minutes at 37°C. Finally, the third step includes the peptide cleanup and elution by means of solid-phase extraction within 60 minutes at room temperature, removing both hydrophilic and hydrophobic contaminations to generate cleanest peptide samples.
The iST technology is displayed by multiple publications in high ranking journals demonstrating both label-free and chemical labeling applications2 for a wide range of sample types. Successful iST processing was demonstrated for cultured, primary or embryonic stem cells, human tissues including rare clinical cancer tissues, or a region- and cell-type resolved quantitative proteomic map of the human heart3, immunoprecipitations, body fluids such as plasma, serum, saliva or urine, as well as various model organisms such as bacteria, yeast or plant samples.

Figure 3: iST 3-step procedure for lysis, digestion and peptide purification.
The method can readily be performed in a 96-well format on robotic liquid handling systems. The PreON platform was built to address the demands of reproducible, robust and sensitive sample preparation (Figure 4). Built-in hardware includes onboard heating shaker, automated centrifuge, gripper, and pipetting units, as well as optical and ultrasonic sensors to control the workflow quality at various steps. The PreON allows research labs to fully automate protein processing at the push of a button. It automates both label-free and chemical labeling workflows using iST technologies and reduces manual processing steps down to five 5 minutes hands-on time. By reducing the technical variability of sample preparation, the PreON platform eliminates both manual processing errors and steps, generating highly reproducible and standardized results (Figure 5).
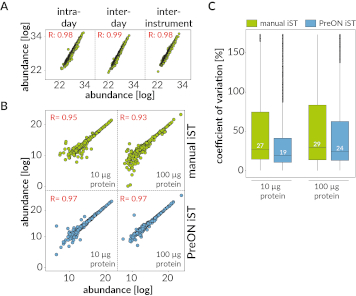
Figure 5: PreON results. (A) Reproducibilities for intra-day, inter-day and inter-instrument sample preparation assessment (yeast pellets, 100 µg protein input). (B) Reproducibilities of manual vs. automated iST sample preparation (human serum, 10 or 100 µg protein input). (C) Technical variability of manual vs. automated iST sample preparation (human serum, 10 or 100 µg protein input).

Figure 4: The PreON platform for automated sample processing for LC-MS/MS-based proteomics.
References
- Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods. 2014 Mar;11(3):319-24. doi: 10.1038/nmeth.2834
- Naamati A, Williamson JC, Greenwood EJ, Marelli S, Lehner PJ, Matheson NJ. Functional proteomic atlas of HIV infection in primary human CD4+ T cells. Elife. 2019 Mar 12;8. pii: e41431. doi: 10.7554/eLife.41431.
- Doll S, Dreßen M, Geyer PE, Itzhak DN, Braun C, Doppler SA, Meier F, Deutsch MA, Lahm H, Lange R, Krane M, Mann M. Region and cell-type resolved quantitative proteomic map of the human heart. Nat Commun. 2017 Nov 13;8(1):1469. doi: 10.1038/s41467-017-01747-2.