State acceptance of the consumption of cannabis and cannabis-derived products for both medical and adult-recreational uses is contingent upon accurate, reliable testing for safety and potency. One of the challenges within this environment is that there are no readily available recognized compendial methods to transform the testing from laboratory-developed methods to validated laboratory methods for general laboratory use. Until such recognized compendial methods are available in the marketplace, it is important to validate methods in the laboratory to demonstrate fitness for intended use.
States’ testing requirements include cannabinoids and terpenoids that are defined by genetics and influenced by environmental factors. As the chemical constituents of cannabis can vary dramatically, accurate analytical data on the chemical content and strength of the product is required for its safe use. Information on the content of potential contaminants also is needed to determine suitability for use. In addition, the analytical characterization of physical factors can play a particularly important role in many different formulations of cannabis-derived products. In many state-regulated cannabis programs, products are analyzed for cannabinoid and terpene profiles, and for the absence of heavy metals, pesticides, and fungal toxins.
ISO/IEC 17025:2017, General requirements for the competence of testing and calibration laboratories, requires that validation be performed for non-standard methods, laboratory-developed methods, or standard methods used outside of their scope. Furthermore, “performance characteristics of validated methods, as assessed for the intended use, shall be relevant to the customers’ needs and consistent with specified requirements.” Because the current methods used for the cannabinoids and terpenes are laboratory-developed, they require validation. Even the U.S. EPA methods frequently used to characterize pesticides or heavy metals are commonly modified for cannabis, so they also require validation.
A related note in ISO/IEC 17025 states: “The techniques used for method validation can be one of, or a combination of, the following: a) calibration or evaluation of bias and precision using reference standards or reference materials;
b) systematic assessment of the factors influencing the result; c) testing method robustness through variation of controlled parameters, such as incubator temperature, volume dispensed; d) comparison of results achieved with other validated methods; e) interlaboratory comparisons; f) evaluation of measurement uncertainty of the results based on an understanding of the theoretical principles of the method and practical experience of the performance of the sampling or test method.”2
Because the standard is written to reflect a wide variety of laboratories, these notes and requirements appear to provide little direction on what specifically is needed for the validation of methods related to the chemical analysis of cannabis and cannabis-derived products. As the medical use of cannabis becomes more prevalent and state accepted, a pharmaceutical approach to method validation would be most appropriate. Adopting a pharmaceutical approach will allow laboratories the use of a readily accepted and standardized approach to method validation for chemical analysis.
Approaches to Validation
When using method validation guidance from organizations such as the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), the U.S. Food and Drug Administration, or USP, it is important to note that there is not a single type of validation that covers all methods. Different analytical performance characteristics are required for the different classifications of analytical methods.
Typical analytical performance characteristics to be considered when using the ICH approach for validation are listed in Table 1.
USP prescribes a similar, yet slightly different approach (Table 2).6 Test requirements vary from exceedingly rigorous analytical determinations to subjective evaluation of qualities. According to USP, considering this broad variety, different test methods require different validation schemes. Different analytical data are needed to determine the fitness for purpose of the analytical method validated. USP categories of methods for validation are as follows:7
- Category I Analytical methods for quantitation of major components of bulk drug substances or active ingredients in finished pharmaceutical products.
- Category II Analytical methods for the determination of impurities in bulk drug substances or degradation compounds in finished pharmaceutical products.
- Category III Analytical methods for determination of performance characteristics.
- Category IV Identification tests.
Quality Control Types Used in Validations
The use of quality controls in validation guarantees that methods of analysis are fit for their intended purpose. In this respect, both systematic errors, leading to bias, and random errors, leading to imprecision, are monitored. To be able to monitor these errors, they should remain constant. Within the laboratory, such constant conditions are typically achieved in one analytical run. Thus, monitoring the precision as an objective of validation does not concern reproducibility or interlaboratory precision, but only repeatability or intra-laboratory precision.
Blanks
Use of various types of blanks enables assessment of how much of the measured signal is attributable to the analyte and how much is to other causes. Types of blank available are:
- Reagent blanks: Reagents used during the analytical process are analyzed to determine whether they contribute to the measurement signal.
- Sample blanks: These are essentially sample matrices with no analyte present, e.g., Cannabis sativa without pesticides present, that give a realistic estimate of interferences that would be encountered in the analysis of test samples.
Routine Test Samples
Routine test samples are useful because of the information they provide on precision, interferences, and other aspects of the test that could realistically be encountered in day-to-day work. If the analyte content of a test material is accurately known, it can be used to assess measurement bias.
Spiked Materials
Spiked materials are substances to which the analyte or analytes of interest have been added. These substances may already contain the analyte of interest, so care is needed to ensure the spiking does not lead to analyte levels outside the working range of the method. Spiking with a known amount of analyte enables the increase in response to the analyte to be measured and calculated in terms of the amount added, even though the absolute amounts of analyte present before and after addition of the spike are not known. Note that most methods of spiking add the analyte in such a way that it will not be as closely bound to the sample matrix as it would be if it was present naturally. Therefore, bias estimates obtained by spiking can be expected to be unrealistic.
Spiking does not necessarily have to be restricted to the analyte of interest. It could include anything added to the sample to gauge the effect of the addition. For example, the sample could be spiked with varying amounts of a particular interference in order to judge the concentration of the interferent at which determination of the analyte is adversely affected.
Reference Materials
It is important to distinguish between reference materials (RMs) and certified reference materials (CRMs) because of the significant difference in how they can be used in method validation. RMs can be any material used as a basis for reference and could include laboratory-prepared standards of known purity. The property or analyte of interest needs to be stable and homogenous, but the material does not need to have the high degree of characterization, metrological traceability, uncertainty, and documentation associated with CRMs.
The characterization of the parameter of interest in a CRM is more strictly controlled than for an RM, and, in addition, the characterized value is certified with documented metrological traceability and uncertainty. Assessment of bias for method validation requires a reliable reference standard, preferably a CRM, with the same matrix and analyte concentrations as the test samples.
Validation Process
Figure 1 illustrates considerations for a validation process to demonstrate the method meets the needs of the end-user and the requirements of ISO/IEC 17025:2017. This is a general process that identifies the aspects to bring method validation to an acceptable closure.
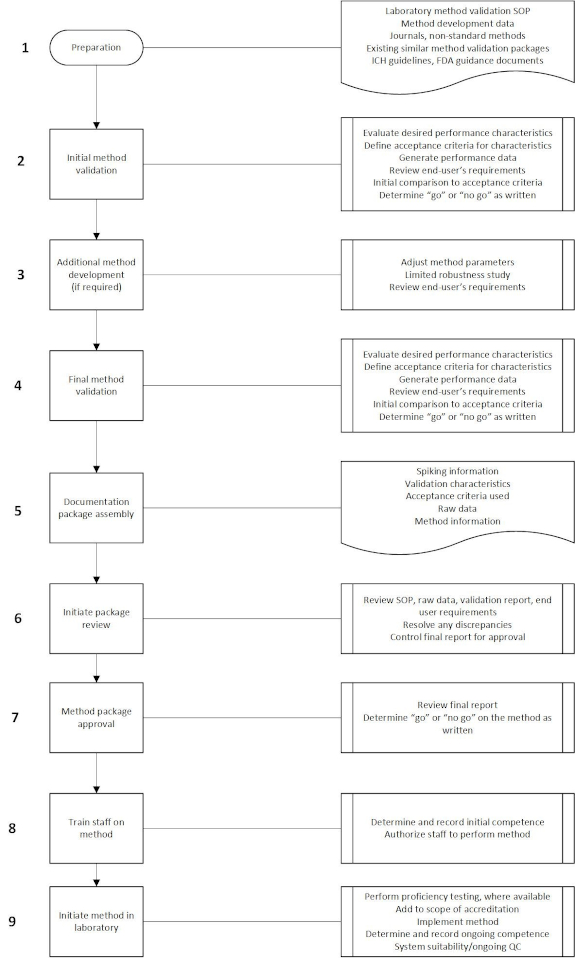
Figure 1. Method Validation Simplified Process Flow
Conclusion
Method validation is indispensable for obtaining reliable results for chemical analysis. The higher the complexity of the method, the more important validation is. Methods related to cannabis testing are notorious for their complexity, on the one hand, because of the instrument itself and, on the other hand, because cannabis and cannabis-related products are complex samples. Therefore, it is important to demonstrate that methods are working as expected and the results obtained are reliable. This information is relevant to both the laboratory and the customer. Method validation is an essential part of good measurement practice because valid data can be produced only when the strengths and weaknesses of a method are understood.
References
- ISO/IEC 17025, General requirements for the competence of testing and calibration laboratories, ISO Geneva, 2017.
- ISO/IEC 17025, General requirements for the competence of testing and calibration laboratories, ISO Geneva, 2017.
- ICH Q2(R1) Validation of Analytical Methods: Text and Methodology, Geneva, incorporated 2005.
- USFDA, Analytical Methods and Methods Validation for Drugs and Biologics Guidance for Industry. Center for Drug Evaluation and Research, Food and Drug Administration, Silver Spring, MD, 2015.
- USP40- NF35, General Chapter 1225, Validation of Compendial Methods, Rockville, MD, 2017.
- USP40-NF35, General Chapter 1225, Validation of Compendial Methods, Rockville, MD, 2017.
- USP40-NF35, General Chapter 1225, Validation of Compendial Methods, Rockville, MD, 2017.
- ISO Guide 30, Reference materials – Selected terms and definitions, ISO Geneva, 2015.