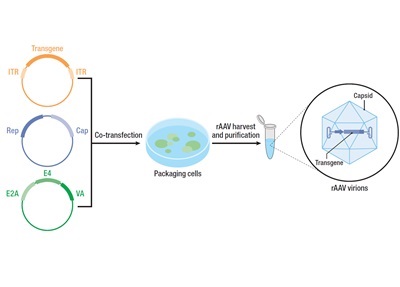
Figure 1. rAAV Vector Design and Assembly for Gene Therapy
by Chelsea B. Pratt, PhD, Biopharma Segment Marketing Manager, Bio-Rad Laboratories and Marwan A. Alsarraj, MS, Associate Director, Marketing Programs, Digital Biology Group, Bio-Rad Laboratories
Viral vectors have played a pivotal role in shaping the gene therapy landscape by leveraging their intrinsic cellular infection capabilities. Notably, adeno-associated viruses (AAVs), characterized by their small size, superior transduction efficiency, low immunogenicity, and potentially tunable tissue specificity, have emerged as the most actively studied carriers for gene therapies. Currently, three AAV therapies have secured approval from the U.S. Food and Drug Administration (FDA) (Luxturna, Zolgensma, and Hemgenix), while another one has received a conditional approval from the European Medicine Agency (EMA) (Rocktavian). However, a large number of pre-clinical and clinical trials are underway to address a wide spectrum of otherwise untreatable genetic disorders.
AAVs, members of the parvovirus family, have not been found to cause disease or be associated with increased immunogenicity, and depend upon adenovirus genes to replicate, hence how they obtained their name. Wild type (WT) AAV consists of single stranded DNA containing two open reading frames (ORFs), Cap (capsid) and Rep (replication), facilitating the integration of the viral genome into a specific human locus establishing latency. Recombinant AAV (rAAV) vectors, engineered for gene therapy, lack 96% of the viral genome, including Rep and Cap genes, preventing replication and integration into the host genome. By retaining only the necessary viral genes to express a recombinant protein-based capsid, rAAVs safely and effectively deliver therapeutic cargo into targeted cells via endocytosis.
Considerations during rAAV Assembly
The success of AAV-based gene therapy hinges on the design of the cargo and capsid. Developing an engineered rAAV vector carrying a transgene involves the use of different plasmids and cell lines for assembly and production. The first plasmid contains the human gene of interest, flanked by inverted terminal repeats (ITRs), and essential DNA sequences facilitating transgene packaging into the vector. The second plasmid is dedicated to carrying specific Rep and Cap sequences, orchestrating the production of AAV capsid proteins. Given the reliance of WT AAV on helper viruses, such as adenoviruses, for replication and infection, a third helper plasmid is employed, supplying E4, E2a, and VA genes facilitating vector reproduction. Co-transfection of these plasmids into host cell lines is the first pivotal step toward generating rAAV vectors (Figure 1). Alternatively, many gene therapy workflows opt for producer lines stably expressing Rep, Cap, and viral helper genes, thereby eliminating the need to for plasmids. Ultimately, a typical fully-formed rAAV vector features a mammalian promoter and ITRs flanking the transgene of interest, enclosed within a viral capsid that dictates tissue specificity.
When designing rAAVs for gene therapy, several other factors need to be considered beyond just the human and viral genes, such as the target cell type, the type of the promoter, and other parameters ensuring the vector’s safety and efficacy. Given these multifaceted considerations, the validation of rAAV assembly and production becomes a meticulous process at each step of the workflow. The challenges encompass not only manufacturing intricacies but also scaling up production and managing costs. This necessitates a continuous refinement of production processes to meet increasing demand.
Addressing Bioprocessing Hurdles in rAAV Production
A critical aspect of gene therapy revolves around viral vector characterization. This involves the implementation of rigorous quality control measures to assess vector yield, purity, and potency. While AAV is normally thought to generate low to no immune response, the vector is generated using biological systems that generate biproducts during production. Measuring and eliminating of bioproducts and contaminants such as host cell proteins (HPC) or host cell DNA (HC DNA), as well as empty or partial capsids, stands as a crucial step to augment infectivity, optimize protein production, and minimize both immunogenicity and off-target effects. The utilization of various analytical techniques proves instrumental in achieving high yields of fully-assembled rAAV particles, thereby amplifying the potency of gene therapy.
rAAV Genome Characterization
During the assembly of capsids in production cell lines, the inadvertent encapsulation of unwanted DNA fragments poses a potential threat to the overall quality of the product. Next-generation sequencing can be used to detect such fragments and verify the accuracy of the genomic material in rAAV. Upon optimization of rAAV particles production, the correct therapeutic dose can be calculated by quantifying viral genome concentration. Reproducible and accurate dosing of any gene therapy product, coupled with the precise viral titration of vector genome copies, is indispensable for determining the optimal transduction efficiency and downstream effectiveness of the therapy.
While quantitative PCR (qPCR) stands as the most established method for nucleic acid quantification, digital PCR (dPCR) has revolutionized the gene therapy field by addressing certain limitations inherent to qPCR. dPCR facilitates absolute quantification and offers higher reproducibility compared to qPCR across both the manufacturing process and purified samples, especially when elevated accuracy and precision are imperative. In contrast to qPCR, dPCR does not rely on a standard curve and perfect DNA amplification efficiency. This is especially important as even with good primer design, the secondary structure in the ITR regions results in PCR efficiencies outside acceptable ranges contributing to high qPCR quantification variability.
Achieving absolute quantification, Droplet Digital PCR (ddPCR) is the gold standard for measuring viral titer within the gene therapy field. During ddPCR nucleic acid molecules are encapsulated in discrete volumetrically defined droplet partitions with yields of 20,000 individual droplets (reactions) in a single 20 µl sample. Following standard PCR conditions, the fluorescence of each droplet is measured to determine the fraction of PCR-positive droplets in the original sample. Subsequently, the data are analyzed to determine the concentration of the target DNA template in the original sample. This technique allows for the simultaneous monitoring of multiple genes to assess genome integrity effectively.
Assessing rAAV Assembly
Validating the identity of viral vector proteins and assessing their integrity and purity during development is a fundamental step to ensuring the safety and efficacy of the final product. The transfection of DNA plasmids, instrumental in generating rAAV vectors, may lead to the production of empty or partially filled vectors devoid of the transgene of interest. The implementation of numerous quality control methods becomes imperative to detect and eliminate such vectors during production, thereby ensuring an acceptable titer of fully assembled rAAV particles and capsid protein subtypes.
Assessment of rAAV assembly, purity, and vector protein identification typically involves Sodium dodecyl sulfate-polyacrylamide (SDS-PAGE) gel followed by electrophoresis and protein staining. For instance, the AAV capsid is comprised of three main proteins, each of which is 87 kD (VP1), 73 kD (VP2), and 61 kD (VP3) in size and are different based on the serotype of the virus. The ratio of these proteins directly influences vector potency by enhancing capsid stability and infectivity. Separation of these proteins by SDS-PAGE electrophoresis allows the determination of VP subtype ratios. However, this is a time-consuming process involving several steps of staining and de-staining. Stain-free Western Blotting workflows offer an alternative solution, expediting the determination of AAV capsid protein ratio.
While gel electrophoresis is a robust tool, its limitation lies in the inability to detect capsid proteins expressed at low levels. Capable of separating and quantifying proteins based on their charge, capillary electrophoresis followed by spectrophotometry is better suited for detecting low abundance proteins. Purity and viral intactness can also be evaluated using either high- performance liquid chromatography (HPLC) or dPCR. Utilizing dPCR allows for the assessment of multiple segments of the viral genome to measure assembly and determining the presence of full or partially full viral capsids.
|
Benefit
|
SDS Page
|
Capillary Electrophoresis
|
qPCR or Digital PCR
|
ELISA
|
HPLC
|
Mass Spectrometry
|
|
Speed or throughput
|
Medium
|
High
|
High
|
Low
|
Medium
|
Low
|
|
Specificity
|
High
|
High
|
High
|
High
|
Low
|
High
|
|
Start-up and running cost
|
Low
|
High
|
Medium
|
Medium
|
High
|
High
|
|
Sensitivity
|
Medium
|
Medium
|
High
|
High
|
Medium
|
High
|
|
Ease of use
|
Easy
|
Medium
|
Medium
|
Hard
|
Medium
|
Hard
|
Transgene expression
The effectiveness of a gene therapy product depends, to a significant extent, on successful transgene expression and the demonstration of intended function. Therefore, it is crucial to assess transgene expression levels in transduced cells throughout the product development process and during lot release. Quantification of in vitro mRNA expression of a transgene is typically achievable using qPCR and/or dPCR, while protein expression can be gauged through immunoassays. Early-stage product development may employ Western blotting or ELISAs to detect protein expression, and at a later stage, flow cytometry can be utilized to validate protein expression on target cell types, which may require multiple markers. When measuring mRNA expression using qPCR or dPCR, a one-step or two-step approach can be taken, with each protocol having its own merits. A one-step RT-PCR combines the reverse transcriptase (RT) reaction and qPCR or dPCR in one tube, allowing for immediate cDNA amplification and rapid data acquisition with minimal interference. On the other hand, a two-step RT-PCR approach separates the RT and qPCR / dPCR steps, allowing for cDNA isolation, which can be utilized in downstream experiments and reserved for any potential audits. While the two-step approach is more efficient and sensitive compared to one-step, it is more time-consuming and may require more optimization. However, confirming not only transgene expression but also protein function post-transduction is essential to ensure the desired therapeutic effect. The methodologies required for this verification will be contingent on the specific mechanism of action of the protein in question.
Conclusion
Addressing bioprocessing challenges in rAAV production is crucial for the success of gene therapy. Robust quality control measures are indispensable to evaluate virus yield, purity, and potency, aiming to eliminate contaminants and incomplete capsids. Innovative techniques, such as next-generation sequencing and dPCR, offer advanced capabilities in verifying genomic material accuracy and absolute quantification of viral titers. With the aim of ensuring effectivity and safety during product manufacturing, the industry could also benefit from collaborating with contract development and manufacturing organizations (CDMOs) or contract research organizations (CROs). Complying with FDA regulatory frameworks on good business practices (e.g. 21 CFR part 11) and adhering to detailed standard operating procedures, can further help ensure product efficacy, purity and ultimately safety for patients.