by Daniel Hornburg, Vice President, Biomarkers and Precision Medicine, Torsten Mueller, Business Development Manager Proteomics, and Stefan Foser, Vice President, Global Pharma, Bruker Daltonics
Mass spectrometry (MS) proteomics has emerged as a promising tool in the field of oncology through its comprehensive study of the biochemical processes underlying cancer. MS allows researchers to study the intricate molecular landscape of tissues, single cells, and individual organelles in humans and model organisms. A deep dive into the proteome is critical to understand the complex, multifaceted nature of cancers. This article explores how recent innovations in MS-based proteomics techniques are opening up new avenues for improving cancer diagnosis and treatment and paving the way for groundbreaking advancements.
The challenge with diagnosis
To diagnose cancer accurately there is no single test; a complete evaluation of a patient’s medical history, physical examination and diagnostic testing is required to determine whether a person has cancer or another infection mimicking its symptoms. Effective testing is used to confirm or eliminate the presence of disease, monitor its progress and plan for and evaluate the efficacy of treatment. Despite billions of dollars being invested into cancer research over many decades, cancer remains one of the biggest challenges to society.
The complexity arises from cancer’s inherently heterogeneous nature. Each type of cancer can present a unique genetic and molecular profile, even within the same tumor type. Additionally, the complex, multi-modal molecular disease pathology of cancer begins with mutations and genomic alterations that occur over time, which can lead to changes in cellular functions, microenvironments, and interactions with other cells. Most of these processes are mediated by proteins and their functionally significant post-translational modifications (PTMs).
It is argued that traditional diagnostics tools are not up to the challenge of capturing the full spectrum of molecular alterations, resulting in incomplete or delayed diagnosis. PTMs can also mean that therapeutics which may be effective for one patient may prove ineffective for another. As a result, there is a critical need for more precise and personalized approaches to understand and combat cancer, highlighting the importance of continued research and innovation in this field.
Exploring cancer-immune system dynamics through immunopeptidomics
Every cell’s deoxyribonucleic acid (DNA) experiences thousands of daily mutations.1 While cellular repair mechanisms fix most of these errors, irreparable cells undergo apoptosis (programmed cell death). Another defense line is the immune system, adept at distinguishing normal cells from those infected by pathogens or transformed into cancer cells. Central to this is the recognition of self and foreign proteins based on immunopeptides presented on the major histocompatibility complex (MHC) molecules on the cell surface. This presentation allows T lymphocytes (T cells) to discern foreign patterns and mount an immune response. Cancer cells not only accumulate mutations that fuel their growth, but also exhibit genomic and proteomic instability, leading to the presentation of new peptides on their surfaces.
While traditional sequencing methods can identify mutations, the actual peptides displayed on cancer cell surfaces can only be determined through MS-based proteomics. The Human Immunopeptidome Project (HIPP) aims to map the entire human immunopeptidome to ultimately make this technology widely accessible in clinical settings. Large-scale quantitative immunopeptidomics could be instrumental in developing cancer immunotherapies and in understanding individual tendencies to common immune diseases and responses to new modalities, including vaccines.2
The development of a high-throughput, sensitive, single-shot MS-based immunopeptidomics workflow leveraging trapped ion mobility spectrometry time-of-flight (TIMS-TOF) technology allows researchers to identify the MHC-I and MHC-II tumor immunopeptidome, aiding in cancer immunotherapy advances.3 This method’s speed, sensitivity, and accuracy has helped create a database containing over 150,000 immune peptides, including both known and novel tumor antigens. Leveraging these comprehensive data, MS-based de novo peptide sequencing algorithms like Novor 2.0 can identify neoantigens from sources such as plasma and extracellular vesicles, even without direct genomics or transcriptomics data. These newly discovered antigens, including rare mutation-derived neoantigens, hold potential as biomarkers or targets for future cancer immunotherapy.4
Revealing cancer insights through spatial proteomics
Spatial biology explores the organization and interactions within tissues at a detailed level, offering profound insights into how cells function within their complex environments. By focusing on the spatial distribution of molecules and cells, spatial biology enhances our understanding of biological processes occurring within tissues and organs. Unlike single-cell next generation sequencing (NGS), which has proven effective in identifying various tumor types and immune states within tumor microenvironments, proteomics enables the analysis of over 10,000 proteins from bulk cellular material.5,6 This advancement in sensitivity and throughput enables researchers to discern proteomic signatures not only across tissues but also at the single-cell level.7,8 Such dynamic spatial information empowers researchers to understand multifaceted biological phenomena and decipher disease progression to pinpoint disease biomarkers, devise therapeutic interventions and generate personalized medicine approaches.
In cancer research, this capability is particularly valuable for studying individual cells assessing immune responses within the tumor microenvironment. For example, gradients of cytokines and chemokines can attract or repel immune cells, ultimately influencing immune infiltration phenotypes relevant to clinical outcomes. Furthermore, profiling extracellular vesicles in tumor environments may uncover biomarkers and shed light on cancer-immune interactions, such as secretion of vesicles containing cancer-specific signatures (e.g., neoantigens) that evade immune responses.
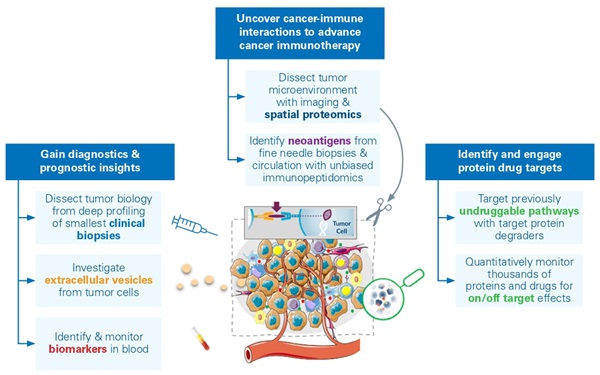 The development of robust and scalable proteomics workflows, capable of analyzing ultra low-input materials, has broad implications in biomedicine. These advancements facilitate early disease detection, as well as the discovery of drug targets and biomarkers, particularly in emerging modalities like CAR-T therapies.9 Enhanced sensitivity of liquid chromatography (LC)-MS workflows, aided by technological advances like TIMS,10 allows for comprehensive and quantitative survey even at the level of single cells.11,12 An emerging application known as deep visual proteomics (DVP) integrates artificial intelligence (AI) for image analysis of cellular characteristics, coupled with automated laser microdissection and LC-MS. In preserved samples of primary melanoma tissue, DVP identified the specific alterations in protein composition caused as normal melanocytes turned into invasive melanoma. These modifications highlight the spatially evolving pathways during cancer progression, including abnormal messenger ribonucleic acid (mRNA) splicing in the growth of metastases, alongside reduced interferon signaling and antigen presentation. The ability for DVPs to maintain precise spatial protein data within tissue samples holds significant promise for molecular analysis in clinical settings.4 This advanced technique enables researchers to examine the tumor microenvironment using small clinical samples like fine needle biopsies, but also allows for the study of other cancer-related factors, such as circulating tumor cells in the blood or secreted proteins and vesicles (Figure 1).
The development of robust and scalable proteomics workflows, capable of analyzing ultra low-input materials, has broad implications in biomedicine. These advancements facilitate early disease detection, as well as the discovery of drug targets and biomarkers, particularly in emerging modalities like CAR-T therapies.9 Enhanced sensitivity of liquid chromatography (LC)-MS workflows, aided by technological advances like TIMS,10 allows for comprehensive and quantitative survey even at the level of single cells.11,12 An emerging application known as deep visual proteomics (DVP) integrates artificial intelligence (AI) for image analysis of cellular characteristics, coupled with automated laser microdissection and LC-MS. In preserved samples of primary melanoma tissue, DVP identified the specific alterations in protein composition caused as normal melanocytes turned into invasive melanoma. These modifications highlight the spatially evolving pathways during cancer progression, including abnormal messenger ribonucleic acid (mRNA) splicing in the growth of metastases, alongside reduced interferon signaling and antigen presentation. The ability for DVPs to maintain precise spatial protein data within tissue samples holds significant promise for molecular analysis in clinical settings.4 This advanced technique enables researchers to examine the tumor microenvironment using small clinical samples like fine needle biopsies, but also allows for the study of other cancer-related factors, such as circulating tumor cells in the blood or secreted proteins and vesicles (Figure 1).
Discovering new drug candidates with chemoproteomics
Proteins are considered to be at the center of all cellular activities, making them prime targets for therapeutic intervention. Scalable strategies have recently emerged to tackle "undruggable" cancer types and other diseases with targeted protein degraders (TPD).13 TPD are a type of therapeutic agent designed to eliminate specific proteins within cells selectively, opening up new possibilities for modulating the abundance and localization of proteins with high therapeutic value. MS plays a crucial role in this process, offering a thorough analysis of cellular proteomes by quantifying over 10,000 proteins. This enables the screening of large compound libraries to observe the effects on the proteome, identifying proteins and pathways that are downregulated (or degraded).
The deep and quantitative profiling of the entire proteome also reveals significant off-target effects, helping researchers to identify the most promising drug candidates. TIMS, combined with parallel accumulation serial fragmentation (PASEF), provides valuable insights into the collisional cross-section (CCS) of molecules.14 This is particularly relevant in activity-based protein profiling (ABPP), where reactive molecules (warheads) target proteins, resulting in distinct masses and unique shape properties that can then be differentiated using TIMS-TOF technology.
Recognizing its significant potential, chemoproteomics has transitioned from academia to industry, with many companies now running programs in preclinical and early clinical development.15 High-throughput proteomics enables the rapid acquisition of complex proteomes, capturing biological phenotypes every few minutes. This allows for the identification of new druggable pathways and the development of therapeutic interventions for complex diseases like cancer. 16,17 Ensuring optimal throughput and workflow robustness is essential for the effective screening of comprehensive compound libraries, with accurate protein identification and sensitive quantification key to reliably determining compound-induced degradation and modification.
Harnessing blood plasma for advanced cancer biomarker detection
Easily collected during routine draws, blood plasma holds a mine of over 100 biomarkers that can be used for early cancer diagnosis.18 A key challenge of using plasma is its wide range of protein concentrations, with a small number dominating the protein mass. However, recent advancements in sample preparation combined with highly efficient MS techniques are enabling plasma proteome analysis.19,20,21 For example, a large cohort lung cancer study demonstrated remarkable sensitivity in detecting lung cancer at various stages using multi-analyte classifiers: 89% for all-stages, 80% for stage I and 98%-100%22 stages III to IV, all at 89% specificity.23
Once protein biomarkers are identified, targeted LC-MS methods like parallel reaction monitoring PASEF (prm-PASEF) can focus in on the proteins and peptides of interest.24 Meanwhile new techniques such as guided data independent acquisition PASEF (g-dia-PASEF) blend targeted and discovery approaches, focusing on known biomarkers while also surveying the entire proteome.25
Blood plasma’s mixed proteome, which consists of secreted proteins, tissue leakage proteins, platelet proteins and potential contaminants, presents a complex but promising area for cancer biomarker discovery.26 Future research might target specific compounds of plasma, such as extracellular vesicles (EVs), which can carry distinct biological insights. Combining de novo sequencing of immunopeptides from circulating or EV-decorating MHC could identify neo cancer antigens. Despite challenges like the lack of sequencing information and the low abundance of MHC peptides, recent innovations in de novo sequencing and highly sensitive LC-MS workflows are clearing the way for new diagnostic opportunities.27
Conclusion
The inherent complexity of cancer biology poses significant challenges for accurate diagnosis and effective treatment. As our understanding of the proteomic landscape of cancer develops, new avenues for early detection, precise classification and personalized therapeutic interventions continue to emerge. These advancements are driven by cutting-edge MS techniques such as the integration of TIMS-TOF mass analyzers, enabling sophisticated acquisition modes like PASEF and novel machine learning algorithms, such as Novor 2.0 to identify cancer neoantigens form the smallest clinical samples. This integration promises to drive new breakthroughs in cancer diagnosis and treatment, ultimately transforming the landscape of cancer care.
References
1. Chatterjee, N.; Walker, G. C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58 (5), 235-263. DOI: 10.1002/em.22087.
2. Vizcaíno, J. A. et al.,The Human Immunopeptidome Project: A Roadmap to Predict and Treat Immune Diseases. Molecular & cellular proteomics. 2020, 19(1), 31–49. https://doi.org/10.1074/mcp.R119.001743.
3. Phulphagar, K. M., Ctortecka, C., et al., Sensitive, high-throughput HLA-I and HLA-II immunopeptidomics using parallel accumulation-serial fragmentation mass spectrometry. Molecular & Cellular Proteomics. 2023, Advance online publication. https://doi.org/10.1016/j.mcpro.2023.100563.
4. Hoenisch Gravel, N., Nelde, A., Bauer, J. et al. TOFIMS mass spectrometry-based immunopeptidomics refines tumor antigen identification. Nat Commun. 2023, 14, 7472. https://doi.org/10.1038/s41467-023-42692-7.
5. Tüshaus, J. et al., A region-resolved proteomic map of the human brain enabled by high-throughput proteomics. EMBO J. 2023, 42 (23), e114665. DOI: 10.15252/embj.2023114665.
6. Mund, A. et al., Deep Visual Proteomics defines single-cell identity and heterogeneity. Nat Biotechnol. 2022, 40(8):1231-1240. DOI: 10.1038/s41587-022-01302-5.
7. Mayer, R. L.; Mechtler, K. Immunopeptidomics in the Era of Single-Cell Proteomics. Biology (Basel). 2023, 12 (12), 1514. DOI: 10.3390/biology12121514.
8. Brunner, A. D. et al., Ultra-high sensitivity mass spectrometry quantifies single-cell proteome changes upon perturbation. Mol. Syst. Biol. 2022, 18 (3), e10798. DOI: 10.15252/msb.202110798.
9. Makhmut, A., Qin, D., Fritzsche, S., Nimo, J., König, J., & Coscia, F. A framework for ultra-low-input spatial tissue proteomics. Methods. 2023, 14(11), 1002-1014.E5. https://doi.org/10.1016/j.cels.2023.10.003.
10. Vasilopoulou, C. G. et al., Trapped ion mobility spectrometry and PASEF enable in-depth lipidomics from minimal sample amounts. Nat. Commun. 2020, 11 (1), 331. DOI: 10.1038/s41467-019-14044-x.
11. Ctortecka, C.; Clark, N. M.; Boyle, B.; Seth, A.; Mani, D. R.; Udeshi, N. D.; Carr, S. A. Automated single-cell proteomics providing sufficient proteome depth to study complex biology beyond cell type classifications. bioRxiv 2024, DOI: 10.1101/2024.01.20.576369.
12. Leduc, A.; Koury, L.; Cantlon, J.; Slavov, N. Massively parallel sample preparation for multiplexed single-cell proteomics using nPOP. bioRxiv 2023, DOI: 10.1101/2023.11.27.568927.
13. Zhao, L.; Zhao, J.; Zhong, K.; et al. Targeted protein degradation: mechanisms, strategies and application. Sig Transduct Target Ther. 2022, 7, 113. DOI: 10.1038/s41392-022-00966-4.
14. Meier, F.; Köhler, N. D.; Brunner, A. D.; Wanka, J. H.; Voytik, E.; Strauss, M. T.; Theis, F. J.; Mann, M. Deep learning the collisional cross sections of the peptide universe from a million experimental values. Nat. Commun. 2021, 12 (1), 1185. DOI: 10.1038/s41467-021-21352-8.
15. Békés, M., Langley, D.R. & Crews, C.M. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov 2022, 21, 181–200. https://doi.org/10.1038/s41573-021-00371-6
16. Messner, C. B., Demichev, V., Wang, Z., Hartl, J., Kustatscher, G., Mülleder, M., & Ralser, M. Mass spectrometry-based high-throughput proteomics and its role in biomedical studies and systems biology. Proteomics, 2023, e2200013. https://doi.org/10.1002/pmic.202200013
17. Kaspar-Schoenefeld, S., Krieger, J. R., Martelli, C., König, A., Hauck, S., Johansson, S., Karger, A., Ohmayer, U., Pecoraro, M., Tenzer, S., Distler, U., Braga-Lagache, S., Strohmidel, P., Abel, L., Schuster, R., Kliewer, G., Kroniger, T., Heikaus, L., Assis, D., Mueller, T., Hornburg, D., High-throughput proteome profiling with low variation in a multi-center study using dia-PASEF, https://doi.org/10.1101/2024.05.29.596405
18. Geyer, P. E.; Holdt, L. M.; Teupser, D.; Mann, M. Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 2017, 13 (9), 942. DOI: 10.15252/msb.20156297.
19. Ferdosi, S. et al., Enhanced Competition at the Nano-Bio Interface Enables Comprehensive Characterization of Protein Corona Dynamics and Deep Coverage of Proteomes. Adv. Mater. 2022, 34 (44), e2206008. DOI: 10.1002/adma.202206008.
20. Viode, A. et al., A simple, time- and cost-effective, high-throughput depletion strategy for deep plasma proteomics. Sci. Adv. 2023, 9 (13), eadf9717. DOI: 10.1126/sciadv.adf9717.
21. Metatla, I. et al., Neat plasma proteomics: getting the best out of the worst. Clin. Proteomics. 2024, 21 (1), 22. DOI: 10.1186/s12014-024-09477-6.
22. Koh, B. et al., Multi-omics profiling with untargeted proteomics for blood-based early detection of lung cancer. medRxiv, 2024.01.03.24300798. https://doi.org/10.1101/2024.01.03.24300798
23. Brzhozovskiy, A. et al., The Parallel Reaction Monitoring-Parallel Accumulation-Serial Fragmentation (prm-PASEF) Approach for Multiplexed Absolute Quantitation of Proteins in Human Plasma. Anal. Chem. 2022, 94 (4), 2016-2022. DOI: 10.1021/acs.analchem.1c03782.
24. Lesur, A.; Bernardin, F.; Koncina, E.; Letellier, E.; Kruppa, G.; Schmit, P. O.; Dittmar, G. Quantification of 782 Plasma Peptides by Multiplexed Targeted Proteomics. J. Proteome Res. 2023, 22 (6), 1630-1638. DOI: 10.1021/acs.jproteome.2c00575.
25. eyer, P. E. et al., Plasma Proteome Profiling to detect and avoid sample-related biases in biomarker studies. EMBO Mol. Med. 2019, 11 (11), e10427. DOI: 10.15252/emmm.201910427.
26. Wahle, M.; Thielert, M.; Zwiebel, M.; Skowronek, P.; Zeng, W. F.; Mann, M. IMBAS-MS Discovers Organ-Specific HLA Peptide Patterns in Plasma. Mol. Cell. Proteomics. 2024, 23 (1), 100689. DOI: 10.1016/j.mcpro.2023.100689.
About the authors
Daniel Horburg is the VP, Biomarkers and Precision Medicine at Bruker Daltonics. Following his Ph.D. under Matthias Mann at the Max Planck Institute, Horburg investigated proteome perturbations in neurodegenerative disorders. He continued his postdoctoral research with Matthias Mann and Felix Meissner, in computational immunoproteomics investigating the communication networks of immune cells and host-pathogen interactions. Later, Horburg joined Mike Snyder’s team at Stanford developing and integrating analytical and computational multi-omics strategies to interrogate metabolic disorders. Outside his academic research, Daniel was working in multiple startups and served as the Vice President Proteomics at Seer before taking on the role at Bruker Daltonics.
Torsten Müller started his path in mass spectrometry-based proteomics in the laboratory of Dr. Dominic Winter at the IBMB in Bonn (DE), working on lysosomal storage disorders. After this initial ignition in LC-MS, he decided to deepen his knowledge in the field by successively joining the labs of Prof. Hanno Steen at the Children’s Hospital in Boston (USA) and Prof. Ruedi Aebersold at ETH in Zürich (CH). Müller obtained his PhD 2020 in the laboratory of Prof. Jeroen Krijgsveld at the University Clinics of Heidelberg and the German Cancer Research Center (DKFZ). In April 2022, Müller joined Bruker Daltonics.
Stefan Foser, currently serving as VP Global Pharma at Bruker Daltonics, combines extensive academic excellence with nearly two decades of professional experience in the pharmaceutical and diagnostics sectors. Holding a Ph.D. in Microbiology, in addition to academic degrees from the Universities of Basel, Switzerland and Mannheim, Germany, and nearly twenty publications and patents, he has been awarded an Executive MBA from the University of St Gallen, Switzerland. Stefan Foser has honed his expertise through pivotal roles at Roche and Siemens Healthineers.