Researchers of phylogenetics and evolution often look at the effects of mutations in the present to predict how those mutations acted in the past and the impact they may have in the future. In order to make these predictions, scientists generally assume that the effects of mutations remain fairly constant over time. However, new research from the University of Chicago suggests this assumption may be wrong for most mutations, and that the effects of mutations steadily change and become less predictable over the course of evolution.
In order to track the influence that certain mutations have had over time, the researchers utilized an advanced combination of techniques recently developed at the university. The study focused on a family of genes that codes for steroid hormone receptors, and the researchers used a massive database of present-day receptor genes to trace the phylogenetic tree backward to the receptors’ common ancestor, dating back approximately 700 million years. The team then reconstructed the sequence of the DNA-binding domain of this ancestor via ancestral sequence reconstruction, as well as those of eight other proteins in the family occurring at various times between that deep ancestor and the present. Expression vectors were synthesized for each of these reconstructions in order to experimentally study their effects in the lab.
The next step of the study was to use a biochemical technique called deep mutational scanning in order to simultaneously measure the effects of massive collections of mutations. The researchers engineered every possible mutation at every site in the sequence of the reconstructed receptors, then incorporated these mutant libraries into yeast cells to study their effects. Fluorescence-activated cell sorting was coupled with sequencing in a technique known as sort-seq in order to measure the ability of every mutant to carry out its biological function. In total, more than 25,000 mutations were tested using this method.
With this huge dataset, the researchers traced how every mutation’s effect changed across the deep evolutionary timeline. The research revealed changes in function due to epistatic interactions, in which a mutation at one site depends on the state of other sites. While the existence of epistatic interactions is well known, the study unexpectedly revealed the prevalence and pervasiveness of the epistatic drift, which was found to cause each mutation to progressively “forget” its previous effects at a largely gradual and steady rate, said Yeonwoo Park, first author of the study. These results revealed not only that predictability decays over time, but that the level of unpredictability can be quantified by performing similar experiments regarding mutations of interest, such as the mutations that give rise to new variants of SARS-CoV-2. This study was published in the journal Science.
“New variants of the virus are constantly arising, and we’d like to predict whether these are likely to be dangerous, or likely to become dangerous with further mutations,” said senior author Joseph Thornton. “Our results mean that a deep mutational scan on one version of the virus can’t produce reliable predictions as the virus gradually drifts away from that old version. But if we have a series of these experiments in the virus across time–like the one we performed for steroid hormone receptors–we’ll know how much the effect of every single mutation is likely to change across a given period of evolutionary time. That means we can express exactly how confident we should be when we predict the effect of any particular mutation in any future virus.”
The team is now expanding their research to characterize not only single mutations, but also every possible pair of mutations that could have occurred in the receptors. These experiments–which would encompass millions rather than tens of thousands of mutations–will enable the researchers to dissect all the interactions that caused epistatic drift during the long-term evolution of the receptor family.
“This new project will reveal the causes of the epistatic drift we discovered in the first study,” said Park. “We expect it to show us how evolution is shaped by the massive set of genetic interactions in a receptor or any other protein–how changes at every single site in the gene alter the biological effect of changes at every other site, and thus rewire its evolutionary potential across time.”
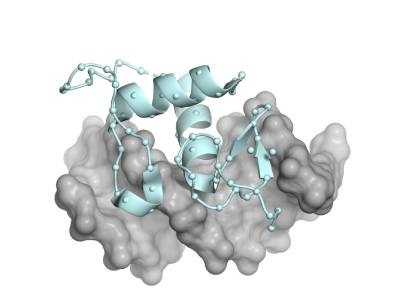
Photo: Rendering of the X-ray crystal structure of the 700-million-year-old ancestral steroid receptor (blue), bound to DNA, gray. Starting from this protein (and eight of its descendants), the Thornton Lab engineered libraries of mutants containing every possible amino acid state at all 76 sites in the protein (spheres). Credit: Joe Thornton, UChicago