Kidney damage is a complication of diabetes, a lifelong condition where the body’s ability to produce or respond to insulin is impaired, resulting in the abnormal metabolism of carbohydrates and elevated blood sugar levels. However, not all people with diabetes develop nephropathy. These individuals with longstanding diabetes who do not develop diabetic nephropathy were studied. Researchers performed an unbiased proteomic analysis of their kidney tissue and identified a unique protein signature. Further research led to the identification of pyruvate kinase M2 (PKM2), a linchpin protein in the pathway that protects these patients. PKM2 is now being investigated as a potential treatment target to prevent or treat diabetic nephropathy.1,2
Proteomics is the critical link between genomics and metabolomics, translating information encoded in nucleic acids such as DNA and RNA into the metabolic processes of living organisms. The large-scale study of the entire complement of proteins and peptides expressed in a cell, tissue or organism is vital for the application of data-driven biology and development of personalized medicines. Elucidating mechanisms of health and disease through proteomics requires fast and reliable analytical methods. However, proteomes are inherently complex and as such, pose huge analytical challenges. A new liquid chromatography-tandem mass spectrometry (LC-MS/MS) method is anticipated to be published that enables the ultrafast analysis of proteomic profiles. The research to develop this technique and high-throughput workflow has been pioneered by Professor Markus Ralser at the Charité university hospital in Berlin, Germany and the Francis Crick Institute in London, in collaboration with a team at SCIEX, Toronto, Canada.3
“Metabolism research has become a hot topic, exerting a major influence on biotechnology, cancer research, immunology, and the rather broad field of metabolic disorders. We have gained access to new technologies over the past few years. We are developing techniques that are capable of accurately measuring metabolic differences in thousands of samples. I think it is now time for us to take the next step and to start transferring our knowledge and technology into clinical practice,” explains Professor Ralser.4
A New Ultrafast Method to Study Proteomics
One approach to reduce the complexity of MS-based proteomic analysis is to separate the sample into fractions before analysis in the mass spectrometer. Although this pre-fractionation approach provides excellent coverage, allowing researchers to investigate even trace amounts of protein or peptide, it is time-consuming and requires extensive resources. Moreover, it can introduce variability between samples and reduce the precision of quantitative measurements. These factors are particularly restrictive when a large number of samples need to be analyzed. For broad proteomic coverage, data-dependent acquisition (DDA) methods are applied. However, this is not absolute and peptides can be missed. An alternative strategy to DDA is that of data-independent acquisition (DIA). DIA methods, such as SWATH Acquisition, are able to provide deep proteome coverage with quantitative precision. With SWATH Acquisition, the mass spectrometer is configured to cycle through a predefined series of wider precursor isolation windows, which fragment in a stepwise, systematic and unbiased fashion all the ionized peptides within the mass range of interest in a sample. In this way. SWATH Acquisition is able to boost the analytical consistency while maintaining relatively high proteome coverage in the measurement of complex biological samples. The use of variable-sized precursor windows, adjusted in size based on known precursor density, is a commonly used way to further boost proteome coverage while maintaining the quality of quantitation.
One way of speeding up an MS analytical run is to use fast chromatographic gradients, but these are limited by the sampling rate of the MS instrument and the occurrence of signal interferences. Short duty cycle times are required to ensure that a sufficient number of data points are sampled for each chromatographic peak to provide accurate quantification. However, to achieve this at fast gradients, the precursor isolation window needs to be broadened, which in turn increases the number of interfering signals. Even on fast quadrupole time-of-flight (TOF) instruments, such as the TripleTOF 6600+ System that can achieve a maximum MS/MS sampling rate of 67 Hz, fast chromatographic gradients require large isolation windows, often ranging from 5 to 30 m/z, in order to accommodate the requisite short cycle times. Such large windows mean that there are more co-eluting precursors being co-fragmented, thus producing more complex spectra as well as signal interferences, which can impact the accuracy of the quantification and the identification of the proteins and peptides.
To address these challenges, an innovative solution has been developed, called Scanning SWATH Acquisition. As MS instruments such as the TripleTOF 6600+ system do not require the accumulation or trapping of ions, acquisition using Q1 windows in a predefined stepwise manner can be replaced by acquisition through a Q1 window that continuously scans across precursor ions in the m/z range of interest (see Figure 1). The fragmentation spectra are then continuously recorded by the TOF analyzer. Thus, Scanning SWATH uncouples the duty cycle time from the isolation window size. These scans also add a new spectral component, which allows the assignation of precursor masses to their corresponding MS/MS spectra. This is possible with the scanning window because the signal corresponding to each fragment from a precursor ion first appears when the leading margin of the window scans over it and disappears when the trailing margin of the window passes over precursor mass. Moreover, a fully automated open-access software suite, DIA-NN, designed for all-in-one DIA data processing has been developed further to enable the processing of Scanning SWATH data. DIA-NN employs deep neural networks (artificial intelligence [AI]) to fully automate the correction for signal interferences and deconvolution of fast DIA data. The software is available as a command line tool but is also equipped with an intuitive simple-to-use graphical user interface. Thus, the Scanning SWATH Acquisition and DIA-NN data processing approach enables DIA at fast chromatographic gradients with ultrafast duty cycles and small isolation window sizes.
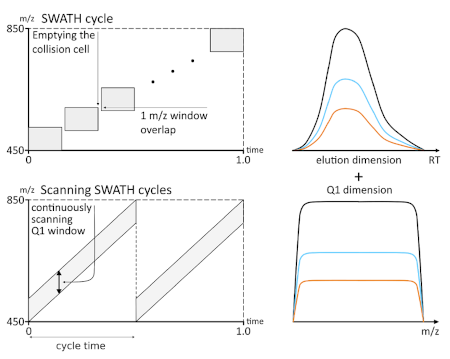
Figure 1: Scanning SWATH Acquisition uncouples duty cycle and isolation window size, and enables the assignment of precursor masses to the MS/MS fragments. Top panel – with conventional SWATH Acquisition, the mass spectrometer repeatedly fragments all the precursor ions within a specified mass range by cycling through a predefined set of relatively large Q1 isolation windows, resulting in fragmentation spectra that are then recorded by the mass analyzer. Bottom panel – with Scanning SWATH, a relatively small Q1 isolation window continuously scans across the precursor mass range of interest, with the mass analyzer continuously recording the fragmentation spectra of the precursors that fall into the window. This effectively uncouples the window size from the cycle time, enabling high selectivity of precursor isolation with ultrafast chromatographic gradients. On both right hand graphs, the x-axis corresponds to the window position (in time or m/z) and y-axis to the signal intensity for each of the fragments, represented with different colors. Image is published under CC-BY and has been obtained from Ref3.3
Raising the Bar for High-Throughput Proteomics
Benchmarking and comparative proteomics experiments using yeast and human cell lysates have demonstrated that Scanning SWATH with DIA-NN provides ultrafast proteomics analysis through shortened duty cycle times and small isolations windows.3 While conventional SWATH Acquisition on a TripleTOF 6600+ System required cycle times in the range of 2–4 seconds and large isolation windows in the range of 15–30 m/z for a 5-minute chromatographic gradient, Scanning SWATH with DIA-NN achieved a cycle time as low as 500 milliseconds with small isolation windows scanning from 450 m/z to 850 m/z.3 It also successfully aligned precursor and MS/MS fragment masses in a chromatogram-type fashion, thereby indicating specific precursor ion each MS/MS fragment trace originated from.
For the LC component of LC-MS, high-flow LC is the standard and preferred method of choice for analytical and industrial disciplines, as it is faster, more reliable, more robust and provides better analytical separation than microflow or nanoflow LC. However, microflow rates are generally used when using fast chromatographic gradients for MS/MS in proteomics because of higher sensitivity of lower flow strategies (i.e. the sample is less diluted). Very fast gradients with high-flow LC requires previously unachievably high MS/MS sampling rates in order to comprehensively cover the proteome. However, Scanning SWATH and DIA-NN have been shown to facilitate the efficient use of fast high-flow LC to provide comprehensive proteome coverage in LC-MS/MS proteomics analysis. Flow rates achieved were as high as 800 μL/min, and the effect of the higher dilution rate could be largely compensated by the higher sample capacity of the high-flow columns, gaining advantage in better separation. 3
High-flow Scanning SWATH and DIA-NN enabled deep proteome coverage at high throughput level that has been previously unattainable in proteomics. With this workflow, 180 samples were measured per day and thousands of unique proteins were quickly and precisely quantified. Compared with other proteomics approaches, high-flow Scanning SWATH and DIA-NN was much faster and had higher throughput, while still recording a high number of data points per peak and precisely quantifying proteins with deep proteome coverage.3
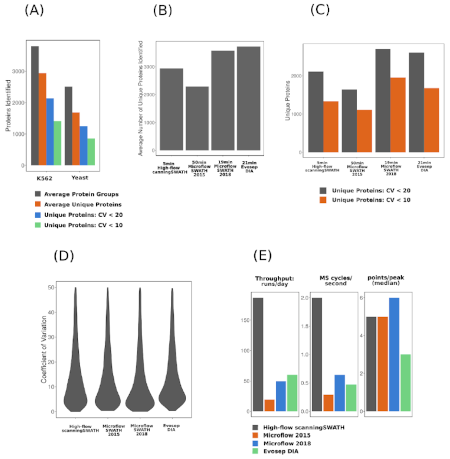
Figure 2: High-flow Scanning SWATH and DIA-NN provides comprehensive protein quantification and is competitive in comparison with methods that are magnitudes lower in throughput. (A) Protein identification with high-flow Scanning SWATH. (B) Identification numbers and quantification performance – average number of unique proteins identified. (C) Average number of unique proteins quantified with coefficient of variation (CV) values below 20% or 10%. (D) CV value distributions. (E) Throughput estimates and the numbers of data points per peak. Image is published under CC-BY and has been obtained from Ref3.3
Ultrafast Proteomics to Accelerate Biomedical Research, Drug Discovery and Development
“From where we were 4–5 years ago and compared with current conventional microflow LC-MS/MS methods, we are now able to perform proteomics up to 5 times faster by using this Scanning SWATH and DIA-NN method. At the same time, the quantitation quality has also improved. Compared with conventional proteomics methods, our new method is 50 times faster. That’s a considerable gain of throughput. At the same time, we have much fewer problems with the LC hardware itself, fewer batch effects, and can run more controls, to better correct for the remaining ones. In fact, we no longer perform any nanoflow LC-MS for proteomics because it is no longer justified in many of our applications” said Professor Ralser.
The combination of Scanning SWATH, DIA-NN advanced data processing, and industry-standard high-flow LC allows the recording of hundreds of precise quantitative proteome maps per day on a single instrument and paves the way for a new generation of cheaper and highly consistent proteomic methods. Such methods are invaluable in the proteomic analysis of clinical samples for therapeutic target identification, screening of compound libraries for drug discovery, and identification of drug effects on the entire proteome of whole cells. These innovative advances should, therefore, empower biomedical scientists to accelerate the elucidation of disease mechanisms, the testing of therapeutic interventions and the development of precision and personalized medicines, such as those aimed at treating microbial infections as well as chronic conditions like diabetes.
Author Information
Dr. Stephen Tate, PhD, is the Senior Research Scientist and Manager of Software, Applications Research at SCIEX, a Danaher operating company and a global leader in the precise quantification of molecules.
References
- Qi W, Keenan HA, Li Q, et al. Nature Medicine 2017, 23, 753.
- Chun N, Wyatt CM, He JC. Kidney International 2017, 92, 780.
- Messner C, Demichev V, Bloomfield N, et al. bioRxiv preprint first posted online May. 31, 2019; doi: http://dx.doi.org/10.1101/656793 (accessed December 2019).
- Charité Press Release – 02.05.2018. Unlocking the secrets of the cell. https://www.charite.de/en/service/press_reports/artikel/detail/den_geheimnissen_im_rezeptbuch_der_zelle_auf_der_spur/ (December 2019).