There has never been a better time to be a cancer researcher. Technological advances have made it possible to analyze tumors at the single-cell level, detect circulating tumor DNA and rare cancer cells in blood samples, and visualize the spatial architecture of a tumor microenvironment. These advances have given researchers a deeper understanding of tumors at the genomic and transcriptomic level, leading to improvements in cancer risk screening, early detection, and precision therapies.
The biggest driver of these advances has been the falling cost of DNA sequencing, enabling the study of cohorts large enough to see statistically meaningful patterns in cancer biology. However, falling costs are not the entire story. More recently, major improvements to sequencing technologies have allowed scientists to generate a more comprehensive view of cancer genomes and transcriptomes, filling in blind spots that previously slowed further progress.
Scientists have deployed these new sequencing technologies to great effect. From characterizing frequently used cancer cell lines to associating patient genotypes with treatment response, the results have been invaluable to the entire cancer research community.
Long-Read Sequencing
One of the most profound ongoing technological shifts has been the development of long-read sequencing. Current sequencing technologies can be grouped into two categories: short-read and long-read sequencers. Short-read sequencers produce reads that are a few hundred bases in length, but the high throughput and low cost per run makes these platforms an attractive option for whole genome sequencing of cancer samples. However, short reads that are just 200 bases long present challenges, including ambiguous mapping, limited phasing, and poor assembly, stemming from the inability to span repetitive regions or SNP deserts that are longer than the read length. Similarly, it is difficult to detect large structural variants from short-read data. If a variant cannot be captured fully in a single read, it can be misrepresented or missed entirely.
Long-read sequencing, on the other hand, generates individual reads that are tens of kilobases in length. One commonly used long-read sequencing technology is single molecule, real-time (SMRT) sequencing. These long reads can fully span even very large structural variants in individual reads, revealing ~3-times more structural variants than short reads with higher precision and recall1. Long reads also enable phasing across SNP deserts to determine whether distant mutations occur in cis or in trans. This information can be pivotal in cases where variants may be acting in concert to affect treatment response. Another powerful application of long reads is discrimination of SNVs in functional genes from those in inactive pseudogenes, which can be important in determining cancer risk status.

Fig 1. The benefits of long-read sequencing
On the transcriptome side, longer reads have made it possible to generate full-length isoform sequences without relying on assembly. Long reads can reveal cryptic exons, retained introns, and other splicing changes that are often cancer-specific and therefore may be missing from the gene models typically used to aid short-read transcript assembly.
Mutation Phasing
Scientists at Memorial Sloan Kettering Cancer Center and other institutes recently reported that SMRT sequencing gave them new insights into why certain patients are “super responders” to alpelisib, a targeted PI3Kα inhibitor2. PIK3CA mutations are present in 40% of ER+ / HER2− primary and metastatic breast cancer tumors, and alpelisib was recently approved by the FDA to treat patients with advanced ER+ breast cancer harboring a PIK3CA mutation.
The team used long-read sequencing of full-length PIK3CA transcripts to look for genetic differences among non-responders, responders, and super-responders. Critical to the success of this approach is the unique ability of SMRT sequencing to generate reads that are both long and highly accurate, delivering fully phased PIK3CA sequences from individual molecules of DNA. The study found that when two mutations occur on the same allele, there is greater susceptibility to PI3Kα inhibitors. When the two mutations are present but located on separate alleles, no such benefit is seen. This explains the super-responder phenotype and provides a clear path to eventually using this information to predict which patients are likely to receive the most benefit from targeted PIK3CA therapy.
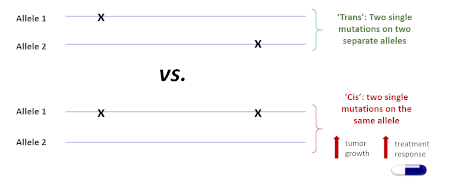
Fig 2. Phasing can reveal hidden cancer biology
In addition, the study revealed that while double mutations in PIK3CA were once believed to be incredibly rare, showing up in less than 1% of cases, SMRT sequencing data revealed that they actually occurred in 12-15% of breast cancers. A review of other data sets suggests that double PIK3CA mutants are more common than previously appreciated in uterine, colorectal, and bladder cancers as well. Routine long-read sequencing could reveal a clinically meaningful number of patients who may derive additional benefit from targeted PIK3CA therapy.
Finally, these results suggest that the method developed by this team might reveal similar patterns of complex mutations in other oncogenes.
Distinguishing Genes from Pseudogenes
Another challenge that long reads can address is discriminating genes from pseudogenes. A classic example is PMS2 genotyping to identify Lynch syndrome. The PMS2 gene has a highly homologous pseudogene, PMS2CL, located on the same chromosome. Thus, a successful cancer risk assay must not only correctly assign SNVs to the gene or pseudogene, but also identify gene conversions and duplications. The standard approach involves combining multiple technologies, and even so, detection of all types of mutations remains unreliable due to frequent sequence exchanges with the pseudogene.
Scientists at Radboud University Medical Center used SMRT sequencing to identify the risk of cancer in Lynch syndrome patients more comprehensively, with a single technology3. Long read sequence data for the entire PMS2 gene was generated in three overlapping long-range amplicons, designed to leverage informative SNPs in PMS2 intron 14 and PMS2CL intron 5 to correctly assign reads to the gene or pseudogene4. With this approach, the team was able to avoid false positives arising from the pseudogene and identify all the variant types that can impact cancer risk.

Fig 3. Targeted sequencing with long reads allows resolution of all types of mutations in the PMS2 gene, yielding more complete information about cancer risk.
Isoform Activity
Beyond DNA, long-read data can give scientists a more comprehensive view of gene activity by generating full-length transcripts. Since no assembly process is needed to piece transcripts together, scientists can have more confidence that the isoforms they detect are real and complete.
In a study from researchers at the Mayo Clinic and other institutions, long-read data was used to analyze truncated isoforms of the androgen receptor, which are associated with resistance to prostate cancer targeted therapies5. Full-length isoform data showed that the known associated variant already being explored as a biomarker for drug resistance, AR-V7, was often co-expressed with another variant, AR-V9. Previously, it was thought that AR-V7 and AR-V9 could be distinguished by their different terminal exons. However, SMRT Sequencing revealed that both isoforms in fact have the same 3’ terminal cryptic exon, and that previous studies had therefore conflated their expression. Long reads allowed scientists to learn that AR-V9, on its own, predicts drug resistance in prostate cancer patients — and likely does so better than the conventionally accepted AR-V7 biomarker.
What’s Next
A common theme across these studies is that long-read sequencing has consistently allowed cancer researchers to make new discoveries. While a decade-plus of short-read data has produced truly exciting information for the cancer research community, there is much more to be learned simply by looking through a different lens. Whether used on its own or in combination with short read data, long reads provide improved access to genetic information.
Going forward, cancer researchers can implement long-read sequencing to find hidden structural variants that affect cancer development or progression, phase haplotypes to more accurately predict cancer risk or treatment response, disambiguate isoforms to provide insights into cancer biology or serve as better biomarkers, and more. New findings from these efforts will continue to transform our understanding of cancer biology and ensure the best chances for identifying novel approaches to predicting, preventing, diagnosing, and treating this disease.
Meredith Ashby, PhD, is the Director of Market Strategy for Microbial Genomics, Cancer and Immunology at Pacific Biosciences. Her mission is to develop PacBio sequencing solutions to meet the unmet needs of scientists seeking to understand all the genomic changes that impact cancer risk, disease progression, treatment response, and relapse.
References
1. Eichler, E. E. (2019). Genetic Variation, Comparative Genomics, and the Diagnosis of Disease. The New England Journal of Medicine, 381(1), 64–74. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/31269367
2. Vasan, N., Razavi, P., Johnson, J. L., Shao, H., Shah, H., Antoine, A., Ladewig, E., et al. (2019). Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3Kα inhibitors. Science, 366(6466), 714–723. Retrieved from https://science.sciencemag.org/content/366/6466/714
3. Variant Analyses of PMS2 by Single-Molecule Long-Read Sequencing, ESHG 2019
K. Neveling, A. Mensenkamp, L. dBruijn, E. Askar, S. vdHeuvel, E. Hoenselaar, R. Derks, M. vdVorst, M. Nelson, L. Vissers, M. Ligtenberg, R. dVoer
4. Neveling, K., Mensenkamp, A., Heiner, C., McLaughlin, I., Harding, J., Aro, L., de Bruijn, L., et al. (2019). The value of long read amplicon sequencing for clinical applications. 69th Annual Meeting of the American Society of Human Genetics. Houston, TX. Retrieved from https://www.pacb.com/wp-content/uploads/Aro-ASHG-2019-The-value-of-long-read-amplicons-for-clinical-applications.pdf
5. Kohli, M., Ho, Y., Hillman, D. W., Van Etten, J. L., Henzler, C., Yang, R., Sperger, J. M., et al. (2017). Androgen receptor variant AR-V9 is co-expressed with AR-V7 in prostate cancer metastases and predicts abiraterone resistance. Clinical Cancer Research, 23(16), 4704–15. Retrieved from http://clincancerres.aacrjournals.org/content/23/16/4704